


Summary
Selective androgen receptor modulators (SARMs) are a class of drugs that selectively activate the androgen receptor in specific tissues, promoting muscle and bone growth while having less effect on male reproductive tissues like the prostate gland.
| Selective androgen receptor modulator | |
|---|---|
| Drug class | |
 Enobosarm (ostarine), a nonsteroidal SARM under investigation for potential medical use. | |
| Class identifiers | |
| Synonyms | Nonsteroidal androgen (although not all SARMs are nonsteroidal)[1] |
| Use | Investigational |
| Biological target | Androgen receptor |
| Chemical class | Mostly nonsteroidal |
| Legal status | |
| Legal status | |
| In Wikidata | |
Non-selective steroidal drugs, called anabolic androgenic steroids (AAS), have been used for various medical purposes, but their side effects limit their use. In 1998, researchers discovered a new class of non-steroidal compounds, the SARMs. These compounds selectively stimulate the androgen receptor, offering potent effects on bone and muscle to increase bone density and lean body mass while having minimal impact on reproductive tissues.
SARMs have been investigated in human studies for the treatment of osteoporosis, cachexia (wasting syndrome), benign prostatic hyperplasia, stress urinary incontinence, and breast cancer. As of 2023[update], there are no SARMs which have been approved by the United States Food and Drug Administration or the European Medicines Agency. Although adverse effects in clinical studies have been infrequent and mild, SARMs can cause elevated liver enzymes, reduction of HDL cholesterol levels, and hypothalamic–pituitary–gonadal axis (HPG axis) suppression, among other side effects.
Since the early twenty-first century, SARMs have been used in doping; they were banned by the World Anti-Doping Agency in 2008. SARMs are readily available on internet-based gray markets and are commonly used recreationally to stimulate muscle growth.
History
editSteroidal androgens
edit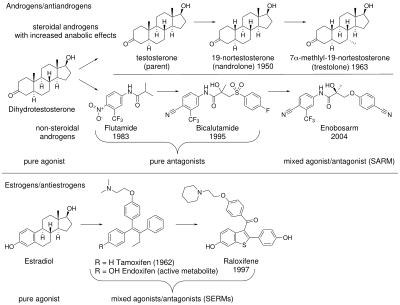

Anabolic androgenic steroids (AAS) are used to treat a variety of medical conditions, but their side effects have fueled a search for a new class of drugs, with a better separation between desirable anabolic and undesirable androgenic effects. The first clinically used AAS was testosterone which was discovered in 1935 and first approved for medical use in 1939.[5] AAS including those produced endogenously such as testosterone and dihydrotestosterone (DHT), bind to and activate the androgen receptor (AR) to produce their effects. AAS effects can be separated into androgenic (the development and maintenance of male sexual characteristics) and anabolic (increasing bone density, muscle mass and strength). AAS also affect hematopoiesis, coagulation, metabolism, and cognition.[6][7] For most medical applications, an AAS with potent anabolic and minimal androgenic and cardiovascular effects would be an advantage.
In the 1930s, 17α-alkylated anabolic steroids were discovered. These have increased metabolic stability and are orally active, but are not tissue selective.[8] These alkylated anabolic steroids still have significant androgenic effects, and are also hepatotoxic.[9][10] In 1950, nandrolone (19-nortestosterone) was first synthesized, which is sometimes considered a SARM due to greater tissue selectivity than testosterone.[8][10][11] In addition, 7α-alkyl substitution of testosterone (for example trestolone) has also been reported to increase its anabolic effects.[8] However, efforts to develop a steroid with anabolic but minimal androgenic effects were not successful.[12]
SERMs
editInterest in nonsteroidal AR mixed agonists/antagonists increased after the therapeutic uses of selective estrogen receptor modulators (SERMs) became evident.[13] The first SERM, tamoxifen, was originally developed as an anti-estrogen contraceptive. However, it was discovered it promoted ovulation in humans by acting as an agonist in ovaries. The drug was then successfully repurposed as a treatment for breast cancer where it was found to act as a full antagonist in breast tissue.[14] Somewhat unexpectedly, it was also discovered that tamoxifen preserves bone density[15] by acting as an agonist in bone resorbing osteoclasts.[16] The clinical success of SERMs stimulated interest in analogous tissue selective drugs that target the AR.[7]
Non-steroidal AR antagonists
editThe chemical starting point for AR mixed agonist/antagonists were nonsteroidal AR antiandrogens such as flutamide, nilutamide, bicalutamide. These antagonists work by binding to the AR to prevent androgenic action; this class of chemicals dates to the 1970s.[6][13] The discovery of arylpropionamides, which share structural similarity with bicalutamide and hydroxyflutamide, suggested a way to make compounds that bind to the AR and produce both anabolic and antiandrogenic effects.[6] Selective androgen receptor modulators (SARMs) were developed out of a desire to maintain the anabolic effects of androgens on muscle and bone, while avoiding side effects on other tissues such as the prostate and cardiovascular system.[9]
Non-steroidal SARMs
editThe first non-steroidal SARMs were developed in 1998 independently by two research groups, one at the University of Tennessee that created an arylpropionamide SARM and Ligand Pharmaceuticals that made a SARM with a quinolone core structure. The name was adopted by analogy with SERMs.[13] Other SARMs include tetrahydroquinolines, tricyclics, bridged tricyclics, aniline, diaryl aniline, bicylclic hydantoins, benzimidazole, imidazolopyrazole, indole, and pyrazoline derivatives.[6] SARMs can be agonists, antagonists, or partial agonists of the AR depending on the tissue, which can enable targeting specific medical conditions while minimizing side effects.[7] Those that have advanced to human trials show stronger effects in bone and muscle tissue and weaker effects in the prostate.[8]
Unlike most current forms of testosterone replacement, SARMs are orally bioavailable[7] and largely eliminated via hepatic metabolism and metabolized through amide hydrolysis in the case of arylpropionamides and A-ring nitro reduction of andarine.[9]
Proposed treatment of hypogonadism
editBecause of the potentially better side effect profile of SARMs compared to testosterone, SARMs have been proposed for use in the treatment of hypogonadism and for androgen replacement therapy.[17][18][19] Phase I and II trials have provided preliminary evidence that the SARMs enobosarm and GSK-2881078 (in elderly men and postmenopausal women), and OPL-88004 (prostate cancer survivors with low levels of testosterone) increase lean body mass and muscle size with little effect on the prostate, supporting the potential of SARMs for use in hormone replacement therapy.[9] However, it has been argued that SARMs are not ideal for use in androgen replacement therapy and could not replace testosterone in this context as they do not reproduce testosterone's full spectrum of effects, including androgenic potentiation via 5α-reduction and aromatization into estrogen.[20][21] Estrogenic signaling in particular is essential for normal male physiology and health, including for instance maintenance of bone strength.[22][23]
Mechanism
editThe mechanism of action of SARMs' tissue-specific effects continues to be debated as of 2020[update].[6][24] A number of hypotheses have been advanced. These include the non-activation of SARMs by 5α-reductase, tissue selective expression of androgen receptor coregulators, non-genomic signaling, and tissue selective uptake of SARMs.[6][25]
5α-Reductase
editTestosterone is active in non-reproductive tissue without activation. In contrast, tissue selective activation by 5α-reductase to the more active form DHT is required for significant activity in reproductive tissue. The net result is that testosterone and its metabolite together are not tissue selective.[26] SARMs are not substrates of 5α-reductase, hence they are not selectively activated like testosterone in tissues such as prostate.[10] This lack of activation effectively imparts a degree of tissue selectivity to SARMs.[27]
Androgen receptor coregulators
editTissue selective transcription coregulator expression is another possible contributor to the selectivity of SARMs.[28][25] Like other type I nuclear receptors, the unliganded androgen receptor (AR) resides in the cytosol complexed with heat shock proteins (HSP). Upon ligand binding, the AR freed from HSPs and translocated into the nucleus where it binds to androgen response elements on DNA to regulate gene expression.[29] AR agonists such as testosterone recruit coactivator proteins to AR that facilitate upregulation of gene expression while antagonists recruit corepressors which down regulate gene expression. Furthermore, the ratio of coactivators to corepressors is known to vary depending on tissue type.[28][19] Structurally, pure AR agonists stabilize the position of helix-12 (H12) in the ligand binding domain of AR near H3 and H4 to produce a surface cleft that binds to a FxxLF motif contained in coactivators.[29] Conversely, antagonists destabilize the agonist conformation of H12 blocking the binding of the FXXLF coactivator motif while facilitating the binding of the corepressor LXX(I/H)IXXX(I/L) motif found in NCOR1 and SMRT corepressors.[29]
In analogy to SERMs, SARMs are mixed agonists/antagonists displaying agonist androgen receptor activity in bone and muscle and partial agonist or antagonist activity in other tissues such as prostate.[25][7] Non-selective agonists such as testosterone are able to recruit coactivators when bound to AR but not corepressors and hence are agonists in all tissues. In contrast, SARMs can recruit both coactivators and corepressors by partially destabilizing the agonist conformation of H12. In tissues where coactivators are in excess (as in bone and muscle), SARMs act as agonists. Conversely, in tissues where corepressors are in excess (such as prostate), SARMs act as partial agonists or antagonists.[25]
In vitro testing of the SARMs enobosarm (ostarine) and YK-11 showed that they bound to the AR, but unlike full AR agonists, they blocked interaction between the N-terminus and C-terminus of AR which resulted in a mixed agonist/antagonist mode of action.[6][25]
Non-genomic signaling
editIn addition to the regulation of gene expression by nuclear AR, membrane associated AR is known to have rapid non-genomic effects on cells through signal transduction cascades. Non-genomic effects appear to significantly contribute to the anabolic effects of androgens whereas genomic effects are primarily responsible for the development of male sexual organs. Furthermore, each steroidal androgen or non-steroidal SARM uniquely influences distinct pathways depending on cell type.[25]
Tissue distribution
editTissue selective uptake into anabolic tissues presents another potential mechanism for SARM tissue selectivity. However autoradiography studies with radiolabeled SARMs show no preferential distribution to anabolic tissues.[10]
Drug candidates
edit| Name | Class | Developer | Investigated for | Highest development stage reached | Structure |
|---|---|---|---|---|---|
| Andarine (S-4, GTx-007) | Arylpropionamide | GTx, Oncternal Therapeutics[31] | Cachexia[31] | Phase I (discontinued)[31] | 
|
| Arcarine (ORM-11984)[32] | Unknown[30] | Orion Corporation[32] | Benign prostatic hyperplasia, hypogonadism, osteoporosis[30] | Phase I (discontinued)[32][30] | Structure undisclosed[30] |
| BMS-564,929 (PS-178990) | Pyrroloimidazole[30] | Bristol-Myers Squibb, Ligand Pharmaceuticals[33] | Andropause, cachexia[33][30] | Phase I (discontinued)[33][34][30] |  [30] [30]
|
| DT-200 (GLPG-0492) | Imidazolidine-2,4-dione | ProSkelia, Akashi Therapeutics, Galapagos NV[35] | Muscular dystrophy, cachexia[35] | Phase I[30][35][36] | 
|
| Enobosarm (ostarine, GTx-024, MK-2866, S-22) | Arylpropionamide | GTx, Veru Healthcare[37] | Breast cancer, cachexia, muscular dystrophy, stress urinary incontinence[37] | Phase III[37][38] | 
|
| GSK-971086 | Indole[39] | GlaxoSmithKline[40] | Sarcopenia[40] | Phase I (discontinued)[40][41] |  [39] [39]
|
| GSK-2849466 | N-arylhydroxyalkyl [42] | GlaxoSmithKline[42] | Cachexia, heart failure[42] | Phase I (discontinued)[42][43] |  [42] [42]
|
| GSK-2881078 | Indole | GlaxoSmithKline[44] | Cachexia[44][45] | Phase II[44][46] | 
|
| GTx-027 | Arylpropionamide | GTx[47][48] | Breast cancer, stress urinary incontinence[47][48] | Phase I (discontinued)[47][48] or preclinical[19] |
|
| LGD-2941 (LGD-122941) | Quinolinone | Ligand Pharmaceuticals[49] | Cachexia, sexual dysfunction, hypogonadism, menopause, osteoporosis[49] | Phase I (discontinued)[49] | 
|
| LGD-4033 (VK5211, ligandrol) | Pyrrolidinebenzonitrile | Ligand Pharmaceuticals[50] | Muscle wasting due to hip fracture, cachexia, hypogonadism, osteoporosis[24][50] | Phase II[50][51] | 
|
| LY305 | N-arylhydroxyalkyl | Eli Lilly[52] | Osteoporosis[52] | Phase I[52] | 
|
| MK-0773 (PF-05314882) | Steroid | GTx, Merck[53] | Sarcopenia, osteoporosis[30][53] | Phase II (discontinued)[30][53][54][55] | 
|
| MK-3984 | Benzylpropionamide | Merck | Sarcopenia[30] | Phase I[30] | 
|
| OPK-88004 (LY-2452473, TT-701) | Indole | Eli Lilly, OPKO[56] | Benign prostatic hyperplasia, quality of life in prostate cancer, erectile dysfunction[56][57] | Phase II[56][58] | 
|
| PF-06260414 | Isoquinoline | Pfizer[59][60] | Cachexia[59] | Phase I (discontinued)[59] | 
|
| Vosilasarm (RAD140, EP0062, testolone) | Phenyloxadiazole | Ellipsis[61] | Breast cancer, osteoporosis, sarcopenia[62] | Phase I/II[62][63] | 
|
| YK-11 | Steroid | Toho University | Muscle wasting[64] | Preclinical | 
|

Certain anabolic steroids, like trestolone, dimethandrolone undecanoate, and 11β-methyl-19-nortestosterone dodecylcarbonate, have also sometimes been classified as SARMs.[30]
Possible therapeutic applications
editDue to their tissue selectivity, SARMs have the potential to treat a wide variety of conditions, including debilitating diseases. They have been investigated in human studies for the treatment of osteoporosis, cachexia, benign prostatic hyperplasia, stress urinary incontinence, prostate cancer, and breast cancer and have also been considered for the treatment of Alzheimer's disease, Duchenne muscular dystrophy, hypogonadism and as a male contraceptive.[19][7] As of 2023[update], there are no SARMs which have been approved for therapeutic use by the United States Food and Drug Administration or the European Medicines Agency.[65]
Most SARMs have been tested in vitro or on rodents, while limited clinical trials in humans have been carried out.[6][66] Initial research focused on muscle wasting.[25] Enobosarm (ostarine) is the most well-studied SARM; according to its manufacturer, GTx Incorporated, 25 studies have been carried out on more than 1,700 humans as of 2020[update] involving doses from 1 to 18 mg each day.[67][24] As of 2020[update], there is little research distinguishing different SARMs from each other.[6] Much of the research on SARMs has been conducted by corporations and has not been made publicly available.[8]
Benign prostatic hyperplasia
editIn rat models of benign prostatic hyperplasia (BPH), a condition where the prostate is enlarged in the absence of prostate cancer, SARMs reduced the weight of the prostate.[66] OPK-88004 advanced to a phase II trial in humans, but it was terminated due to difficulty in measuring prostate size, the trial's primary endpoint.[19]
Cancer
editSARMs may help treat AR and estrogen receptor (ER) positive breast cancer, which comprise the majority of breast cancers.[7][68] AAS were historically used successfully to treat AR positive breast cancer, but were phased out after the development of antiestrogen therapies, due to androgenic side effects and concerns about aromatization to estrogen (which does not occur with SARMs).[68][25] Although a trial on AR positive triple negative breast cancer (which is ER-) was ended early due to lack of efficacy, enobosarm showed benefits in some patients with ER+, AR+ breast cancer in a phase II study. In patients with more than 40 percent AR positivity as determined by immunohistochemistry, the clinical benefit rate (CBR) was 80 percent and the objective response rate (ORR) was 48 percent—which was considered promising given that the patients had advanced disease and had been heavily pretreated.[69][68] In 2022, the FDA granted fast track designation to enobosarm for AR+, ER+, HER2- metastatic breast cancer.[70] Other SARMs such as vosilasarm have reached clinical trials in breast cancer patients.[61]
Bone and muscle wasting
editAs of 2020[update], there are no drugs approved to treat muscle wasting in people with chronic diseases, and there is therefore an unmet need for anabolic drugs with few side effects. One aspect hindering drug approval for treatments for cachexia and sarcopenia (two types of muscle wasting) is disagreement in what outcomes would demonstrate the efficacy of a drug. Several clinical trials have found that SARMs improve lean mass in humans, but it is not clear whether strength and physical function are also improved. After promising results in a phase II trial, a phase III trial of enobosarm was proven to increase lean body mass but did not show significant improvement in function. It and other drugs have been refused regulatory approval due to a lack of evidence that they increased physical performance; preventing decline in functionality was not considered an acceptable endpoint by the Food and Drug Administration. It is not known how SARMs interact with dietary protein intake and resistance training in people with muscle wasting.[24][19]
Phase II trials of enobosarm for stress urinary incontinence—considered promising, given that the levator ani muscle in the pelvic floor has a high androgen receptor density—did not meet their endpoint and were abandoned.[19][25]
Unlike other treatments for osteoporosis, which work by decreasing bone loss, SARMs have shown potential to promote growth in bone tissue. LY305 showed promising results in a phase I trial in humans.[19]
Side effects
editIn contrast to AAS and testosterone replacement, which have many side effects that have curtailed their medical use, SARMs are well tolerated and have mild and infrequent adverse events in randomized controlled trials.[66] SARMs are sometimes claimed to be non-virilizing (non-masculinizing).[19][71] In actuality however, SARMs are largely uncharacterized clinically in terms of potential virilizing effects.[6] In addition, SARMs cannot be aromatized to estrogen, thus causing no estrogenic side effects, for instance gynecomastia.[72][19][7]
SARM use can cause elevated liver enzymes and reduction in HDL cholesterol.[72][19] Transdermal administration via a skin patch may reduce these effects.[19][52] Several case reports have associated SARMs with hepatocellular drug-induced liver injury when used recreationally,[73] it is not known if the risk is significant for medical use.[66][7] Whether SARMs increase the risk of cardiovascular events is unknown.[66][7] SARMs have less effect on blood lipid profiles than testosterone replacement; it is not known whether androgen-induced HDL reductions increase cardiovascular risk; and SARMs increase insulin sensitivity and lower triglycerides.[7][24]
Although they cause less suppression of the hypothalamic–pituitary–gonadal axis (HPG axis) than testosterone, studies have found that gonadotropins, free and total testosterone, and SHBG can be reduced in a compound- and dose-dependent fashion in men from SARM usage.[6][24] Typically SHBG is reduced along with total testosterone and total cholesterol while hematocrit is increased. Most studies have found that follicle-stimulating hormone (FSH), luteinizing hormone (LH), prostate-specific antigen, estradiol, and DHT levels are not altered.[66] Of SARMs that have been investigated, enobosarm is one of the least suppressive of gonadotropins, even in doses much higher than used in clinical trials. How the HPG axis is affected in women using SARMs is unknown.[6][24] SARMs' effect in suppressing the gonadotropins FSH and LH is what makes SARMs potentially useful as a male contraceptive.[74]
Non-medical use
editOutside of pharmaceutical research, SARMs are a gray market substance produced by small laboratories and often marketed as a research chemical supposedly not for human consumption.[6][75][76] Marketing SARMs for human consumption is illegal in some jurisdictions and has led to criminal convictions in the United States[77] and the largest-ever fine levied under Australia's Therapeutic Goods Act 1989.[78] Although SARMs are readily available for purchase on the internet, one study found that a majority of products advertised as SARMs online were mislabeled. Anecdotes and guides on usage can also be found online and on social media.[79][72][7] Some compounds are commonly marketed for recreational use as SARMs despite having a different mechanism of action. These substances include ibutamoren (MK-677), which increases secretion of growth hormone; GW501516 (cardarine), an exercise mimetic that works as an agonist of the PPARβ/δ; and SR9009 (Stenabolic), an agonist of the Rev-Erb, which plays a role in circadian rhythm.[6][80]
SARMs are used by bodybuilders and competitive athletes due to their anabolic and lack of androgenic effects,[7] particularly in the United States, Europe, and other western countries.[72] Some individuals using SARMs recreationally combine multiple SARMs or take a SARM along with other compounds, although there is no research on combining SARMs. The doses used often exceed those from clinical trials; nevertheless, the fat-free mass gained from SARMs is generally lower than what is obtained with moderate doses of testosterone derivatives.[6] According to one study of SARM users, more than 90 percent were satisfied with their usage and 64 percent would take SARMs again even though a majority experienced adverse effects.[81]
SARMs were banned by the World Anti-Doping Agency (WADA) in 2008.[6] SARMs can be detected in urine and hair after consumption.[82] WADA reported its first adverse analytical finding for SARMs in 2010 and the number of positive tests has increased since then; the most commonly detected SARMs are enobosarm (ostarine) and LGD-4033 (ligandrol).[83][84] Athletes competing in the NFL, NBA, UFC, NCAA, and the Olympics have tested positive.[73] There is limited evidence on how SARMs affect athletic performance.[85]
Terminology
editSARMs are sometimes also referred to as "nonsteroidal androgens",[1][86] although not all SARMs are nonsteroidal in structure and steroidal SARMs also exist.[30] The first SARMs, discovered in 1998, were nonsteroidal and were initially called nonsteroidal androgens.[87] In 1999, the term "selective androgen receptor modulator" or "SARM" was introduced, as the mixed agonist–antagonist and tissue-selective activity of these nonsteroidal androgen receptor agonists had similarities with selective estrogen receptor modulators (SERMs).[17] Despite its widespread use, the term "selective androgen receptor modulator" has been criticized by some authors, like David Handelsman, who argue that it is a misleading pharmaceutical marketing term rather than an accurate pharmacological description.[20] He has also critiqued notions that SARMs isolate anabolic effects from androgenic or virilizing effects, as has been previously claimed in the case of anabolic steroids.[20][88][89][90]
References
edit- ^ a b Tauchen J, Jurášek M, Huml L, Rimpelová S (February 2021). "Medicinal Use of Testosterone and Related Steroids Revisited". Molecules. 26 (4): 1032. doi:10.3390/molecules26041032. PMC 7919692. PMID 33672087.
SARMs are a novel group of compounds developed to selectively augment anabolic effects in muscles and bones, while avoiding undesirable androgenic effects in skin, larynx, and reproductive organs. The majority of these compounds lack the structural functionalities of the original anabolic steroids and are sometimes termed nonsteroidal androgens. It was hoped that these agents could be used in cases where conventional anabolic steroids produced undesirable side-effects, such as virilization in women and prostate hyperplasia in men [67]. Despite the enormous effort that has been expended in the development of selective anabolic agents, the androgenic effect is very hard to remove completely and many of the currently developed SARMs still do have some androgenic activity.
- ^ Koh B (22 March 2013). "Anti-doping agency warns cheats on the health risks of Endurobol". The Conversation. Archived from the original on 4 September 2023. Retrieved 4 September 2023.
- ^ Mohler ML, Sikdar A, Ponnusamy S, Hwang DJ, He Y, Miller DD, et al. (February 2021). "An Overview of Next-Generation Androgen Receptor-Targeted Therapeutics in Development for the Treatment of Prostate Cancer". International Journal of Molecular Sciences. 22 (4): 2124. doi:10.3390/ijms22042124. PMC 7924596. PMID 33672769.
- ^ de Vera IM, Waaninayake US, Burris TP (October 2018). "Structural Insights into Estrogen Receptors and Antiestrogen Therapies". In Zhang X (ed.). Estrogen Receptor and Breast Cancer: Celebrating the 60th Anniversary of the Discovery of ER. Springer International Publishing. ISBN 978-3-319-99350-8.
- ^ Brannigan RE (2021). "Testosterone Therapy and Male Fertility". In Mulhall JP, Maggi M, Trost L (eds.). Controversies in Testosterone Deficiency. Springer International Publishing. pp. 57–70 (57). doi:10.1007/978-3-030-77111-9_6. ISBN 978-3-03-077111-9. S2CID 237993949.
- ^ a b c d e f g h i j k l m n o p Machek SB, Cardaci TD, Wilburn DT, Willoughby DS (2020). "Considerations, possible contraindications, and potential mechanisms for deleterious effect in recreational and athletic use of selective androgen receptor modulators (SARMs) in lieu of anabolic androgenic steroids: A narrative review". Steroids. 164: 108753. doi:10.1016/j.steroids.2020.108753. ISSN 0039-128X. PMID 33148520. S2CID 225049089. Archived from the original on 2023-08-30. Retrieved 2023-08-30.
Sex-specific SARM effects on humans also remain considerably nebulous. SARMs may represent a more tempting option for female recreational use given potential previous tendencies towards less androgenic AAS (i.e. oxandrolone) [103]. Regardless, as the latter still imposes risk for permanent masculinization and hepatotoxicity, SARMs are largely uncharacterized for female-specific impacts.
- ^ a b c d e f g h i j k l m Solomon ZJ, Mirabal JR, Mazur DJ, Kohn TP, Lipshultz LI, Pastuszak AW (2019). "Selective Androgen Receptor Modulators: Current Knowledge and Clinical Applications". Sexual Medicine Reviews. 7 (1): 84–94. doi:10.1016/j.sxmr.2018.09.006. PMC 6326857. PMID 30503797.
- ^ a b c d e Jasuja R, Zacharov MN, Bhasin S (2012). "The state-of-the-art in the development of selective androgen receptor modulators". In Nieschlag E, Behre HM (eds.). Testosterone: Action, Deficiency, Substitution (4th ed.). Cambridge University Press. pp. 459–460. doi:10.1017/CBO9781139003353.022. ISBN 978-1-107-01290-5.
- ^ a b c d Bhasin S, Krishnan V, Storer TW, Steiner M, Dobs AS (2023). "Androgens and Selective Androgen Receptor Modulators to Treat Functional Limitations Associated With Aging and Chronic Disease". The Journals of Gerontology: Series A. 78 (Supplement_1): 25–31. doi:10.1093/gerona/glad027. PMC 10272983. PMID 37325955. Archived from the original on September 1, 2023. Retrieved September 1, 2023.
- ^ a b c d Bhasin S, Jasuja R (2009). "Selective Androgen Receptor Modulators (SARMs) as Function Promoting Therapies". Current Opinion in Clinical Nutrition and Metabolic Care. 12 (3): 232–240. doi:10.1097/MCO.0b013e32832a3d79. ISSN 1363-1950. PMC 2907129. PMID 19357508.
- ^ Thevis M, Schänzer W (2010). "Synthetic Anabolic Agents: Steroids and Nonsteroidal Selective Androgen Receptor Modulators". Doping in Sports. Handbook of Experimental Pharmacology. Vol. 195. pp. 99–126. doi:10.1007/978-3-540-79088-4_5. ISBN 978-3-540-79087-7. PMID 20020362.
One of the first synthetic analogs to testosterone prepared by the Noble laureate Ruzicka was 17α-methyltestosterone (Ruzicka et al. 1935).
- ^ Katzung BG (2017). Basic and Clinical Pharmacology (14th ed.). McGraw Hill Professional. p. 741. ISBN 978-1-259-64116-9.
- ^ a b c d Temerdashev AZ, Dmitrieva EV (2020). "Methods for the Determination of Selective Androgen Receptor Modulators". Journal of Analytical Chemistry. 75 (7): 835–850. doi:10.1134/S1061934820070187. S2CID 220398030.
- ^ Quirke VM (2017). "Tamoxifen from Failed Contraceptive Pill to Best-Selling Breast Cancer Medicine: A Case-Study in Pharmaceutical Innovation". Frontiers in Pharmacology. 8: 620. doi:10.3389/fphar.2017.00620. PMC 5600945. PMID 28955226.
- ^ Jordan VC (November 2021). "Turning scientific serendipity into discoveries in breast cancer research and treatment: a tale of PhD students and a 50-year roaming tamoxifen team". Breast Cancer Research and Treatment. 190 (1): 19–38. doi:10.1007/s10549-021-06356-8. PMC 8557169. PMID 34398352.
- ^ Komm BS, Cheskis B, Bodine PV (November 2007). "Regulation of Bone Cell Function by Estrogens". In Marcus R (ed.). Osteoporosis. Elsevier Science. pp. 383–412 (409). ISBN 978-0-08-055347-4. Archived from the original on 2024-03-08. Retrieved 2024-02-04.
- ^ a b Negro-Vilar A (October 1999). "Selective androgen receptor modulators (SARMs): a novel approach to androgen therapy for the new millennium". The Journal of Clinical Endocrinology and Metabolism. 84 (10): 3459–3462. doi:10.1210/jcem.84.10.6122. PMID 10522980.
We have chosen the term selective androgen receptor modulators (SARMs) after the terminology currently used for similar molecules targeting the estrogen receptor. ... Desired profile of activity of new SARMs: male applications: Selected indications may include glucocorticoid-induced osteoporosis, androgen replacement in elderly men, HIV-wasting, cancer cachexia, certain anemias, muscular dystrophies, and male contraception.
- ^ Zaveri NT, Murphy BJ (2007). "Nuclear hormone receptors". In Taylor JB, Triggle DJ (eds.). Comprehensive Medicinal Chemistry II. Elsevier. pp. 993–1036. doi:10.1016/B0-08-045044-X/00063-8. ISBN 9780080450445.
A SARM for the treatment of hypogonadism or osteoporosis would be an AR agonist in the muscle and bone, with minimal hypertrophic agonist effects in the prostate.
- ^ a b c d e f g h i j k l Christiansen AR, Lipshultz LI, Hotaling JM, Pastuszak AW (March 2020). "Selective androgen receptor modulators: the future of androgen therapy?". Translational Andrology and Urology. 9 (Suppl 2): S135–S148. doi:10.21037/tau.2019.11.02. ISSN 2223-4683. PMC 7108998. PMID 32257854.
- ^ a b c Handelsman DJ (July 2022). "History of androgens and androgen action". Best Practice & Research. Clinical Endocrinology & Metabolism. 36 (4): 101629. doi:10.1016/j.beem.2022.101629. PMID 35277356.
The next invention was that of the first non-steroidal androgen by Dalton et al. [111] in 1998, six decades after the first non-steroidal estrogen [112]. This creates a new class of non-steroidal synthetic androgen, often termed Specific Androgen Receptor Modulators (SARM), a misleading marketing term rather than an accurate pharmacological description [113,114], usurping a speculative but unsound analogy with Specific Estrogen Receptor Modulators (SERM). [...] none of the non-steroidal androgens under development [116,117] are marketed by 2021. Yet hope springs eternal for this new attempt to separate anabolic from androgenic properties of androgens to facilitate marketing for muscle wasting and other selective effects of testosterone.
- ^ Feingold KR, Anawalt B, Blackman MR, Boyce A, Chrousos G, Corpas E, et al. (5 October 2020). "Androgen Physiology, Pharmacology, Use and Misuse". Endotext. PMID 25905231.
These features suggest that non-steroidal androgens have potential for development into pharmacologic androgen therapy regimens as tissue-selective mixed or partial androgen agonists ("selective androgen receptor modulators", SARM) (419, 718). Conversely, they are not ideal for androgen replacement therapy where the full spectrum of testosterone effects including aromatization is idealy required, especially for tissues such as the brain (148, 159) and bone (153) where aromatization is a prominent feature of testosterone action.
- ^ Russell N, Grossmann M (July 2019). "Mechanisms in Endocrinology: Estradiol as a male hormone". Eur J Endocrinol. 181 (1): R23–R43. doi:10.1530/EJE-18-1000. PMID 31096185.
- ^ Cooke PS, Nanjappa MK, Ko C, Prins GS, Hess RA (July 2017). "Estrogens in Male Physiology". Physiol Rev. 97 (3): 995–1043. doi:10.1152/physrev.00018.2016. PMC 6151497. PMID 28539434.
- ^ a b c d e f g Fonseca GW, Dworatzek E, Ebner N, Von Haehling S (2020). "Selective androgen receptor modulators (SARMs) as pharmacological treatment for muscle wasting in ongoing clinical trials". Expert Opinion on Investigational Drugs. 29 (8): 881–891. doi:10.1080/13543784.2020.1777275. PMID 32476495. S2CID 219174372.
- ^ a b c d e f g h i Narayanan R, Coss CC, Dalton JT (April 2018). "Development of selective androgen receptor modulators (SARMs)". Molecular and Cellular Endocrinology. 465: 134–142. doi:10.1016/j.mce.2017.06.013. PMC 5896569. PMID 28624515.
- ^ Kicman AT (June 2008). "Pharmacology of anabolic steroids". British Journal of Pharmacology. 154 (3): 502–521. doi:10.1038/bjp.2008.165. PMC 2439524. PMID 18500378.
- ^ Gao W, Dalton JT (February 2007). "Ockham's razor and selective androgen receptor modulators (SARMs): are we overlooking the role of 5α-reductase?". Molecular Interventions. 7 (1): 10–3. doi:10.1124/mi.7.1.3. PMC 2040232. PMID 17339601.
- ^ a b Smith CL, O'Malley BW (February 2004). "Coregulator function: a key to understanding tissue specificity of selective receptor modulators". Endocrine Reviews. 25 (1): 45–71. doi:10.1210/er.2003-0023. PMID 14769827.
- ^ a b c Tan MH, Li J, Xu HE, Melcher K, Yong EL (January 2015). "Androgen receptor: structure, role in prostate cancer and drug discovery". Acta Pharmacologica Sinica. 36 (1): 3–23. doi:10.1038/aps.2014.18. PMC 4571323. PMID 24909511.
- ^ a b c d e f g h i j k l m n o p Xie Y, Tian Y, Zhang Y, Zhang Z, Chen R, Li M, et al. (February 2022). "Overview of the development of selective androgen receptor modulators (SARMs) as pharmacological treatment for osteoporosis (1998-2021)". Eur J Med Chem. 230: 114119. doi:10.1016/j.ejmech.2022.114119. PMID 35063736. S2CID 245941791.
- ^ a b c "Andarine". AdisInsight. Springer Nature Switzerland AG. Archived from the original on 2024-01-03. Retrieved 2024-01-03.
- ^ a b c "Arcarine". Synapse. PatSnap. Archived from the original on 2024-01-14. Retrieved 2024-01-14.
- ^ a b c "PS 178990". AdisInsight. Springer Nature Switzerland AG. Archived from the original on 2024-01-14. Retrieved 2024-01-14.
- ^ "PS-178990". Synapse. PatSnap. Archived from the original on 2024-01-14. Retrieved 2024-01-14.
- ^ a b c "DT 200". AdisInsight. Springer Nature Switzerland AG. Archived from the original on 2024-01-03. Retrieved 2024-01-14.
- ^ "GLPG0492 Intervention/Treatment Search Results". ClinicalTrials.gov. U.S. National Library of Medicine. Archived from the original on 2024-02-08. Retrieved 2024-02-08.
- ^ a b c "Enobosarm - Veru Healthcare". AdisInsight. Springer Nature Switzerland AG. Archived from the original on 2023-04-05. Retrieved 2024-01-14.
- ^ "Enobosarm Intervention/Treatment Search Results". ClinicalTrials.gov. U.S. National Library of Medicine. Archived from the original on 2024-03-08. Retrieved 2024-02-11.
- ^ a b "GSK-971086". Inxight Drugs. National Center for Advancing Translational Sciences (NCATS), U.S. National Institutes of Health.
- ^ a b c "GSK 971086". AdisInsight. Springer Nature Switzerland AG. Archived from the original on 2024-01-14. Retrieved 2024-01-14.
- ^ "GSK971086 Intervention/Treatment Search Results". ClinicalTrials.gov. U.S. National Library of Medicine. Archived from the original on 2024-03-08. Retrieved 2024-02-11.
- ^ a b c d e "GSK 2849466". AdisInsight. Springer Nature Switzerland AG. Archived from the original on 2024-01-14. Retrieved 2024-01-14.
- ^ "GSK2849466 Intervention/Treatment Search Results". ClinicalTrials.gov. U.S. National Library of Medicine. Archived from the original on 2024-03-08. Retrieved 2024-02-11.
- ^ a b c "GSK 2881078". AdisInsight. Springer Nature Switzerland AG. Archived from the original on 2024-01-03. Retrieved 2024-01-14.
- ^ Mohan D, Rossiter H, Watz H, Fogarty C, Evans RA, Man W, et al. (1 March 2023). "Selective androgen receptor modulation for muscle weakness in chronic obstructive pulmonary disease: a randomised control trial". Thorax. 78 (3): 258–266. doi:10.1136/thorax-2021-218360. ISSN 0040-6376. PMC 9985744. PMID 36283827. Archived from the original on 2 September 2023. Retrieved 11 September 2023.
- ^ "GSK2881078 Intervention/Treatment Search Results". ClinicalTrials.gov. U.S. National Library of Medicine. Archived from the original on 2024-03-08. Retrieved 2024-02-11.
- ^ a b c "Delving into the Latest Updates on GTx-027 with Synapse". Synapse. 13 October 2024. Retrieved 22 October 2024.
- ^ a b c "Research programme: selective androgen receptor modulators". AdisInsight. 16 April 2020. Retrieved 22 October 2024.
- ^ a b c "LGD 2941". AdisInsight. Springer Nature Switzerland AG. Archived from the original on 2024-01-14. Retrieved 2024-01-14.
- ^ a b c "VK 5211". AdisInsight. Springer Nature Switzerland AG. Archived from the original on 2024-01-03. Retrieved 2024-01-07.
- ^ "VK5211 Intervention/Treatment Search Results". ClinicalTrials.gov. U.S. National Library of Medicine. Archived from the original on 2024-03-08. Retrieved 2024-02-11.
- ^ a b c d Krishnan V, Patel NJ, Mackrell JG, Sweetana SA, Bullock H, Ma YL, et al. (2018). "Development of a selective androgen receptor modulator for transdermal use in hypogonadal patients". Andrology. 6 (3): 455–464. doi:10.1111/andr.12479. PMID 29527831. S2CID 3858281.
- ^ a b c "MK 0773". AdisInsight. Springer Nature Switzerland AG. Archived from the original on 2023-07-09. Retrieved 2024-01-14.
- ^ Papanicolaou DA, Ather SN, Zhu H, Zhou Y, Lutkiewicz J, Scott BB, et al. (2013). "A phase IIA randomized, placebo-controlled clinical trial to study the efficacy and safety of the selective androgen receptor modulator (SARM), MK-0773 in female participants with sarcopenia". J Nutr Health Aging. 17 (6): 533–43. doi:10.1007/s12603-013-0335-x. PMID 23732550. S2CID 42439768.
- ^ "MK0773 Intervention/Treatment Search Results". ClinicalTrials.gov. U.S. National Library of Medicine. Archived from the original on 2024-02-08. Retrieved 2024-02-08.
- ^ a b c "OPK 88004". AdisInsight. Springer Nature Switzerland AG. Archived from the original on 2024-01-03. Retrieved 2024-01-14.
- ^ Pencina KM, Burnett AL, Storer TW, Guo W, Li Z, Kibel AS, et al. (2021). "A Selective Androgen Receptor Modulator (OPK-88004) in Prostate Cancer Survivors: A Randomized Trial". The Journal of Clinical Endocrinology & Metabolism. 106 (8): 2171–2186. doi:10.1210/clinem/dgab361. PMC 8277210. PMID 34019661.
- ^ "OPK-88004 Intervention/Treatment Search Results". ClinicalTrials.gov. U.S. National Library of Medicine. Archived from the original on 2024-03-08. Retrieved 2024-02-11.
- ^ a b c "PF 626414". AdisInsight. Springer Nature Switzerland AG. Archived from the original on 2024-01-14. Retrieved 2024-01-14.
- ^ "PF-06260414 Intervention/Treatment Search Results". ClinicalTrials.gov. U.S. National Library of Medicine.
- ^ a b Lim E, Hamilton E, Palmieri C, Arkenau HT, Brook S, Fisher G, et al. (1 March 2023). "Abstract OT1-02-02: A phase 1/2 study to evaluate the safety and efficacy of EP0062, an oral Selective Androgen Receptor Modulator (SARM), for the treatment of AR+/HER2-/ER+ advanced breast cancer". Cancer Research. 83 (5_Supplement): OT1–02–02-OT1-02-02. doi:10.1158/1538-7445.SABCS22-OT1-02-02. S2CID 257320030.
- ^ a b "Vosilasarm - Ellipses Pharma". AdisInsight. Springer Nature Switzerland AG. Archived from the original on 2024-01-03. Retrieved 2024-01-12.
- ^ "RAD140 Intervention/Treatment Search Results". ClinicalTrials.gov. U.S. National Library of Medicine.
- ^ Lee SJ, Gharbi A, Shin JE, Jung ID, Park YM (2021). "Myostatin inhibitor YK11 as a preventative health supplement for bacterial sepsis". Biochemical and Biophysical Research Communications. 543: 1–7. doi:10.1016/j.bbrc.2021.01.030. ISSN 0006-291X. PMID 33588136. S2CID 231938058.
- ^ Leciejewska N, Jędrejko K, Gómez-Renaud VM, Manríquez-Núñez J, Muszyńska B, Pokrywka A (February 2024). "Selective androgen receptor modulator use and related adverse events including drug-induced liver injury: Analysis of suspected cases". European Journal of Clinical Pharmacology. 80 (2): 185–202. doi:10.1007/s00228-023-03592-3. PMC 10847181. PMID 38059982.
- ^ a b c d e f Sigalos JT, Walker DT, Lipschultz LI (2023). "Selective Androgen Receptor Modulators in the Treatment of Hypogonadism and Men's Health". Men's Reproductive and Sexual Health Throughout the Lifespan: An Integrated Approach to Fertility, Sexual Function, and Vitality. Cambridge University Press. p. 266. doi:10.1017/9781009197533.034. ISBN 978-1-009-19755-7.
- ^ Zajac JD, Seeman E, Russell N, Ramchand SK, Bretherton I, Grossmann M, et al. (2020). "Testosterone". Encyclopedia of Bone Biology. Academic Press. p. 545. ISBN 978-0-12-814082-6.
- ^ a b c Dai C, Ellisen LW (2023). "Revisiting Androgen Receptor Signaling in Breast Cancer". The Oncologist. 28 (5): 383–391. doi:10.1093/oncolo/oyad049. PMC 10166165. PMID 36972361. Archived from the original on 2023-10-12. Retrieved 2023-09-10.
- ^ Palmieri C, Linden HM, Birrell S, Lim E, Schwartzberg LS, Rugo HS, et al. (2021). "Efficacy of enobosarm, a selective androgen receptor (AR) targeting agent, correlates with the degree of AR positivity in advanced AR+/estrogen receptor (ER)+ breast cancer in an international phase 2 clinical study". Journal of Clinical Oncology. 39 (15_suppl): 1020. doi:10.1200/JCO.2021.39.15_suppl.1020. ISSN 0732-183X. S2CID 236407030. Archived from the original on 2023-10-12. Retrieved 2023-09-10.
- ^ "FDA Grants Fast Track Designation to Enobosarm in AR+, ER+, HER2- Metastatic Breast Cancer". Cancer Network. 10 January 2022. Archived from the original on 27 August 2023. Retrieved 27 August 2023.
- ^ Zhang X, Sui Z (February 2013). "Deciphering the selective androgen receptor modulators paradigm". Expert Opin Drug Discov. 8 (2): 191–218. doi:10.1517/17460441.2013.741582. PMID 23231475. S2CID 2584722.
- ^ a b c d Xie Y, Tian Y, Zhang Y, Zhang Z, Chen R, Li M, et al. (15 February 2022). "Overview of the development of selective androgen receptor modulators (SARMs) as pharmacological treatment for osteoporosis (1998–2021)". European Journal of Medicinal Chemistry. 230: 114119. doi:10.1016/j.ejmech.2022.114119. ISSN 0223-5234. PMID 35063736. S2CID 245941791.
- ^ a b Hahamyan H, Gould H, Gregory A, Dodson C, Gausden E, Voos J, et al. (2023). "Poster 390: Systematic Review of SARMs Abuse in Athletes". Orthopaedic Journal of Sports Medicine. 11 (7_suppl3): 2325967123S00352. doi:10.1177/2325967123S00352. ISSN 2325-9671. PMC 10392554. S2CID 260375399.
- ^ Bhasin S (2015). "Selective Androgen Receptor Modulators as Function Promoting Therapies". The Journal of Frailty & Aging. 4 (3): 121–122. doi:10.14283/jfa.2015.65. ISSN 2260-1341. PMC 6039107. PMID 27030938.
- ^ Sobolevsky T, Ahrens B (2021). "High-throughput liquid chromatography tandem mass spectrometry assay as initial testing procedure for analysis of total urinary fraction". Drug Testing and Analysis. 13 (2): 283–298. doi:10.1002/dta.2917. ISSN 1942-7603. PMID 32852861. S2CID 221347916. Archived from the original on 2023-09-01. Retrieved 2023-09-01.
- ^ Turnock DL, Gibbs DN (2023). "Click, click, buy: The market for novel synthetic peptide hormones on mainstream e-commerce platforms in the UK". Performance Enhancement & Health. 11 (2): 100251. doi:10.1016/j.peh.2023.100251. ISSN 2211-2669. S2CID 257706930.
- ^ Oberheiden N (26 June 2023). "The FDA Continues to Crack Down on SARM Manufacturing and Distribution". Federal Lawyer. Archived from the original on 4 November 2023. Retrieved 13 October 2023.
- ^ Jacobson HL, Zhang A, Forrai Z (3 August 2021). "Failure to remove unlawful advertising attracts $12 million penalty". Lexology. Archived from the original on 4 November 2023. Retrieved 13 October 2023.
- ^ Hahamyan HA, Vasireddi N, Voos JE, Calcei JG (2023). "Social media's impact on widespread SARMs abuse". The Physician and Sportsmedicine. 51 (4): 291–293. doi:10.1080/00913847.2022.2078679. ISSN 0091-3847. PMID 35574698. S2CID 248812455.
- ^ Handschin C (2016). "Caloric restriction and exercise mimetics: Ready for prime time?". Pharmacological Research. 103: 158–166. doi:10.1016/j.phrs.2015.11.009. PMC 4970791. PMID 26658171.
- ^ Efimenko IV, Valancy D, Dubin JM, Ramasamy R (2022). "Adverse effects and potential benefits among selective androgen receptor modulators users: a cross-sectional survey". International Journal of Impotence Research. 34 (8): 757–761. doi:10.1038/s41443-021-00465-0. ISSN 1476-5489. PMID 34471228. S2CID 237378326.
- ^ Kintz P, Ameline A, Gheddar L, Raul JS (2019). "LGD-4033, S-4 and MK-2866 – Testing for SARMs in hair: About 2 doping cases". Toxicologie Analytique et Clinique. 31 (1): 56–63. Bibcode:2019ToxAC..31...56K. doi:10.1016/j.toxac.2018.12.001. ISSN 2352-0078. Archived from the original on 2023-08-30. Retrieved 2023-08-30.
- ^ "Selektive Androgenrezeptor-Modulatoren (SARMs)". Institut für Biochemie, Deutsche Sporthochschule Köln (in German). Archived from the original on 1 September 2023. Retrieved 1 September 2023.
- ^ Kintz P (5 January 2022). "The forensic response after an adverse analytical finding (doping) involving a selective androgen receptor modulator (SARM) in human athlete". Journal of Pharmaceutical and Biomedical Analysis. 207: 114433. doi:10.1016/j.jpba.2021.114433. ISSN 1873-264X. PMID 34715583. S2CID 240229684.
- ^ Warrier AA, Azua EN, Kasson LB, Allahabadi S, Khan ZA, Mameri ES, et al. (2023). "Performance-Enhancing Drugs in Healthy Athletes: An Umbrella Review of Systematic Reviews and Meta-analyses". Sports Health: A Multidisciplinary Approach. 16 (5): 695–705. doi:10.1177/19417381231197389. ISSN 1941-7381. PMC 11346223. PMID 37688400. S2CID 261620672. Archived from the original on 2023-11-04. Retrieved 2023-10-12.
- ^ Brown TR (December 2004). "Nonsteroidal selective androgen receptors modulators (SARMs): designer androgens with flexible structures provide clinical promise". Endocrinology. 145 (12): 5417–9. doi:10.1210/en.2004-1207. PMID 15545403.
- ^ Dalton JT, Mukherjee A, Zhu Z, Kirkovsky L, Miller DD (March 1998). "Discovery of nonsteroidal androgens". Biochem Biophys Res Commun. 244 (1): 1–4. doi:10.1006/bbrc.1998.8209. PMID 9514878.
- ^ Handelsman DJ (May 2011). "Commentary: androgens and "anabolic steroids": the one-headed janus". Endocrinology. 152 (5): 1752–4. doi:10.1210/en.2010-1501. PMID 21511988.
Although development of the first nonsteroidal androgens (17, 18) as candidate selective AR modulators (19) raises hope of resurrecting this defunct term (20), prereceptor activation mechanisms cannot apply to nonsteroidal androgens, and the singular AR lacks a dual drive mechanism of the other paired sex steroid receptors. Consequently, it is not surprising that available knowledge (21) provides only slender hope that this failed, and probably false, dichotomy will now succeed through a renewed search guided by the same in vivo bioassay.
- ^ Handelsman DJ (July 2021). "Androgen Misuse and Abuse". Endocr Rev. 42 (4): 457–501. doi:10.1210/endrev/bnab001. PMID 33484556.
However, a third major quest, for the development of a nonvirilizing androgen ("anabolic steroid") suitable for use in women and children, based on dissociating the virilizing from the anabolic effects of androgens failed comprehensively (36). This failure is now understood as being due to the discovery of a singular androgen receptor (AR) together with the misinterpretation of nonspecific whole animal androgen bioassays employed to distinguish between anabolic and virilizing effects (37). The term "androgen" is used herein for both endogenous and synthetic androgens including references to chemicals named elsewhere as "anabolic steroids," "anabolic-androgenic steroids," or "specific AR modulators" (SARM), which continue to make an obsolete and oxymoronic distinction between virilizing and anabolic effects of androgens where there is no difference (36).
- ^ Handelsman DJ (2012-07-26). "Androgen therapy in non-gonadal disease". Testosterone. Cambridge University Press. pp. 372–407. doi:10.1017/cbo9781139003353.018. ISBN 978-1-139-00335-3.
The development of nonsteroidal androgens, marketed as "selective androgen receptor modulators" (SARMs), offers new possibilities for adjuvant pharmacological androgen therapy. In contrast to the full spectrum of androgen effects of testosterone, such SARMs would be pure androgens not subject to tissue-specific activation by aromatization to a corresponding estrogen or to amplification of androgenic potency by 5α-reduction. In this context the endogenous pure androgens nandrolone and DHT can be considered prototype SARMs. SARMs are not the modern embodiment of so-called "anabolic steroids," an outdated term referring to hypothetical but nonexistent non-virilizing androgens targeted exclusively to muscle, a failed concept lacking biological proof of principle (Handelsman 2011).