


Summary
In the Arrhenius model of reaction rates, activation energy is the minimum amount of energy that must be available to reactants for a chemical reaction to occur.[1] The activation energy (Ea) of a reaction is measured in kilojoules per mole (kJ/mol) or kilocalories per mole (kcal/mol).[2] Activation energy can be thought of as the magnitude of the potential barrier (sometimes called the energy barrier) separating minima of the potential energy surface pertaining to the initial and final thermodynamic state. For a chemical reaction to proceed at a reasonable rate, the temperature of the system should be high enough such that there exists an appreciable number of molecules with translational energy equal to or greater than the activation energy. The term "activation energy" was introduced in 1889 by the Swedish scientist Svante Arrhenius.[3]

Other uses edit
Although less commonly used, activation energy also applies to nuclear reactions[4] and various other physical phenomena.[5][6][7]
Temperature dependence and the relation to the Arrhenius equation edit
The Arrhenius equation gives the quantitative basis of the relationship between the activation energy and the rate at which a reaction proceeds. From the equation, the activation energy can be found through the relation

where A is the pre-exponential factor for the reaction, R is the universal gas constant, T is the absolute temperature (usually in kelvins), and k is the reaction rate coefficient. Even without knowing A, Ea can be evaluated from the variation in reaction rate coefficients as a function of temperature (within the validity of the Arrhenius equation).
At a more advanced level, the net Arrhenius activation energy term from the Arrhenius equation is best regarded as an experimentally determined parameter that indicates the sensitivity of the reaction rate to temperature. There are two objections to associating this activation energy with the threshold barrier for an elementary reaction. First, it is often unclear as to whether or not reaction does proceed in one step; threshold barriers that are averaged out over all elementary steps have little theoretical value. Second, even if the reaction being studied is elementary, a spectrum of individual collisions contributes to rate constants obtained from bulk ('bulb') experiments involving billions of molecules, with many different reactant collision geometries and angles, different translational and (possibly) vibrational energies—all of which may lead to different microscopic reaction rates.[citation needed]
Catalysts edit
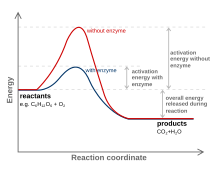


A substance that modifies the transition state to lower the activation energy is termed a catalyst; a catalyst composed only of protein and (if applicable) small molecule cofactors is termed an enzyme. A catalyst increases the rate of reaction without being consumed in the reaction.[8] In addition, the catalyst lowers the activation energy, but it does not change the energies of the original reactants or products, and so does not change equilibrium.[9] Rather, the reactant energy and the product energy remain the same and only the activation energy is altered (lowered).
A catalyst is able to reduce the activation energy by forming a transition state in a more favorable manner. Catalysts, by nature, create a more "comfortable" fit for the substrate of a reaction to progress to a transition state. This is possible due to a release of energy that occurs when the substrate binds to the active site of a catalyst. This energy is known as Binding Energy. Upon binding to a catalyst, substrates partake in numerous stabilizing forces while within the active site (e.g. hydrogen bonding or van der Waals forces). Specific and favorable bonding occurs within the active site until the substrate forms to become the high-energy transition state. Forming the transition state is more favorable with the catalyst because the favorable stabilizing interactions within the active site release energy. A chemical reaction is able to manufacture a high-energy transition state molecule more readily when there is a stabilizing fit within the active site of a catalyst. The binding energy of a reaction is this energy released when favorable interactions between substrate and catalyst occur. The binding energy released assists in achieving the unstable transition state. Reactions without catalysts need a higher input of energy to achieve the transition state. Non-catalyzed reactions do not have free energy available from active site stabilizing interactions, such as catalytic enzyme reactions.[10]
Relationship with Gibbs energy of activation edit
In the Arrhenius equation, the term activation energy (Ea) is used to describe the energy required to reach the transition state, and the exponential relationship k = A exp(−Ea/RT) holds. In transition state theory, a more sophisticated model of the relationship between reaction rates and the transition state, a superficially similar mathematical relationship, the Eyring equation, is used to describe the rate constant of a reaction: k = (kBT / h) exp(−ΔG‡ / RT). However, instead of modeling the temperature dependence of reaction rate phenomenologically, the Eyring equation models individual elementary steps of a reaction. Thus, for a multistep process, there is no straightforward relationship between the two models. Nevertheless, the functional forms of the Arrhenius and Eyring equations are similar, and for a one-step process, simple and chemically meaningful correspondences can be drawn between Arrhenius and Eyring parameters.
Instead of also using Ea, the Eyring equation uses the concept of Gibbs energy and the symbol ΔG‡ to denote the Gibbs energy of activation to achieve the transition state. In the equation, kB and h are the Boltzmann and Planck constants, respectively. Although the equations look similar, it is important to note that the Gibbs energy contains an entropic term in addition to the enthalpic one. In the Arrhenius equation, this entropic term is accounted for by the pre-exponential factor A. More specifically, we can write the Gibbs free energy of activation in terms of enthalpy and entropy of activation: ΔG‡ = ΔH‡ − T ΔS‡. Then, for a unimolecular, one-step reaction, the approximate relationships Ea = ΔH‡ + RT and A = (kBT/h) exp(1 + ΔS‡/R) hold. Note, however, that in Arrhenius theory proper, A is temperature independent, while here, there is a linear dependence on T. For a one-step unimolecular process whose half-life at room temperature is about 2 hours, ΔG‡ is approximately 23 kcal/mol. This is also the roughly the magnitude of Ea for a reaction that proceeds over several hours at room temperature. Due to the relatively small magnitude of TΔS‡ and RT at ordinary temperatures for most reactions, in sloppy discourse, Ea, ΔG‡, and ΔH‡ are often conflated and all referred to as the "activation energy".
The enthalpy, entropy and Gibbs energy of activation are more correctly written as Δ‡Ho, Δ‡So and Δ‡Go respectively, where the o indicates a quantity evaluated between standard states.[11][12] However, some authors omit the o in order to simplify the notation.[13][14]
The total free energy change of a reaction is independent of the activation energy however. Physical and chemical reactions can be either exergonic or endergonic, but the activation energy is not related to the spontaneity of a reaction. The overall reaction energy change is not altered by the activation energy.
Negative activation energy edit
In some cases, rates of reaction decrease with increasing temperature. When following an approximately exponential relationship so the rate constant can still be fit to an Arrhenius expression, this results in a negative value of Ea.
Elementary reactions exhibiting negative activation energies are typically barrierless reactions, in which the reaction proceeding relies on the capture of the molecules in a potential well. Increasing the temperature leads to a reduced probability of the colliding molecules capturing one another (with more glancing collisions not leading to reaction as the higher momentum carries the colliding particles out of the potential well), expressed as a reaction cross section that decreases with increasing temperature. Such a situation no longer leads itself to direct interpretations as the height of a potential barrier.[15]
Some multistep reactions can also have apparent negative activation energies. For example, the overall rate constant k for a two-step reaction A ⇌ B, B → C is given by k = k2K1, where k2 is the rate constant of the rate-limiting slow second step and K1 is the equilibrium constant of the rapid first step. In some reactions, K1 decreases with temperature more rapidly than k2 increases, so that k actually decreases with temperature corresponding to a negative observed activation energy.[16][17][18]
An example is the oxidation of nitric oxide which is a termolecular reaction . The rate law is with a negative activation energy.[19][20] This is explained by the two-step mechanism: and .

![{\displaystyle v=k\,\left[{\rm {NO}}\right]^{2}\,\left[{\rm {O_{2}}}\right]}](https://wikimedia.org/api/rest_v1/media/math/render/svg/ae77cc1d5a0ef31cc0ac2b83fbb421dcca3d9e68)


Certain cationic polymerization reactions have negative activation energies so that the rate decreases with temperature. For chain-growth polymerization, the overall activation energy is , where i, p and t refer respectively to initiation, propagation and termination steps. The propagation step normally has a very small activation energy, so that the overall value is negative if the activation energy for termination is larger than that for initiation. The normal range of overall activation energies for cationic polymerization varies from 40 to 60 kJ/mol.[21]

See also edit
References edit
- ^ "Activation Energy". www.chem.fsu.edu. Archived from the original on 2016-12-07. Retrieved 2017-01-13.
- ^ Espenson, James (1995). Chemical Kinetics and Reaction Mechanisms. McGraw-Hill. ISBN 0070202605.
- ^ "Activation Energy and the Arrhenius Equation – Introductory Chemistry- 1st Canadian Edition". opentextbc.ca. Archived from the original on 2017-07-08. Retrieved 2018-04-05.
- ^ Kagan, Harris; Barrett, Tom. "Energy in a Modern Society: XIV. Nuclear energy" (Course). Ohio State University. Retrieved 15 October 2021.
- ^ Wang, Jenqdaw; Raj, Rishi (1990). "Estimate of the Activation Energies for Boundary Diffusion from Rate-Controlled Sintering of Pure Alumina, and Alumina Doped with Zirconia or Titania". Journal of the American Ceramic Society. 73 (5): 1172. doi:10.1111/j.1151-2916.1990.tb05175.x.
- ^ Kiraci, A; Yurtseven, H (2012). "Temperature Dependence of the Raman Frequency, Damping Constant and the Activation Energy of a Soft-Optic Mode in Ferroelectric Barium Titanate". Ferroelectrics. 432: 14–21. doi:10.1080/00150193.2012.707592. S2CID 121142463.
- ^ Terracciano, Anthony C; De Oliveira, Samuel; Vazquez-Molina, Demetrius; Uribe-Romo, Fernando J; Vasu, Subith S; Orlovskaya, Nina (2017). "Effect of catalytically active Ce 0.8 Gd 0.2 O 1.9 coating on the heterogeneous combustion of methane within MgO stabilized ZrO 2 porous ceramics". Combustion and Flame. 180: 32–39. doi:10.1016/j.combustflame.2017.02.019.
- ^ "General Chemistry Online: FAQ: Chemical change: What are some examples of reactions that involve catalysts?". antoine.frostburg.edu. Retrieved 2017-01-13.
- ^ Bui, Matthew. "The Arrhenius Law: Activation Energies". Chemistry LibreTexts. UC Davis. Retrieved February 17, 2017.
- ^ Berg, Jeremy (2019). Biochemistry - Ninth Edition. New York, NY: WH Freeman and Company. pp. 240–244. ISBN 978-1-319-11467-1.
- ^ "Enthalpy of activation". IUPAC Gold Book (2nd edition, on-line version). IUPAC (International Union of Pure and Applied Chemistry). 2019. Retrieved 10 May 2020.
- ^ Steinfeld, Jeffrey I.; Francisco, Joseph S.; Hase, William L. (1999). Chemical Kinetics and Dynamics (2nd ed.). Prentice Hall. p. 301. ISBN 0-13-737123-3.
- ^ Atkins, Peter; de Paula, Julio (2006). Atkins' Physical Chemistry (8th ed.). W.H.Freeman. p. 883. ISBN 0-7167-8759-8.
... but we shall omit the standard state sign to avoid overburdening the notation.
- ^ Laidler, Keith J.; Meiser, John H. (1982). Physical Chemistry. Benjamin/Cummings. p. 381. ISBN 0-8053-5682-7.
- ^ Mozurkewich, Michael; Benson, Sidney (1984). "Negative activation energies and curved Arrhenius plots. 1. Theory of reactions over potential wells". J. Phys. Chem. 88 (25): 6429–6435. doi:10.1021/j150669a073.
- ^ Espenson, James H. (1981). Chemical kinetics and reaction mechanisms. McGraw-Hill. p. 121. ISBN 0-07-019667-2.
- ^ Atkins, Peter; de Paula, Julio (2006). Atkins' Physical Chemistry (8th ed.). W. H. Freeman. p. 822. ISBN 0-7167-8759-8.
- ^ Kadir, Tamara (10 September 2020). "3.2.2: Pre-equilibrium Approximation". Chem Libre Texts. Retrieved 23 January 2022.
The overall activation energy is negative if Ea1 + Ea2 < Ea−1
- ^ Moore, John W.; Pearson, Ralph G. (1981). Kinetics and mechanism (3rd ed.). John Wiley and Sons. p. 316. ISBN 0-471-03558-0.
- ^ Engel, Thomas; Reid, Philip (2006). Physical Chemistry. Pearson. Benjamin-Cummings. p. *34. ISBN 0-8053-3842-X.
- ^ Cowie, J. M. G. (1991). Polymers: Chemistry and Physics of Modern Materials (2nd ed.). Blackie (USA: Chapman & Hall+). p. 88. ISBN 0-216-92980-6.