


Summary
Boroxine (B3H3O3) is a 6-membered heterocyclic compound composed of alternating oxygen and singly-hydrogenated boron atoms. Boroxine derivatives (boronic anhydrides) such as trimethylboroxine and triphenylboroxine also make up a broader class of compounds called boroxines.[1] These compounds are solids that are usually in equilibrium with their respective boronic acids at room temperature.[1][2][3] Beside being used in theoretical studies, boroxine is primarily used in the production of optics.[4]

| |

| |
| Names | |
|---|---|
| IUPAC name
1,3,5,2,4,6-Trioxatriborinane
| |
| Other names
cyclotriboroxane
| |
| Identifiers | |
| |
3D model (JSmol)
|
|
| ChemSpider |
|
PubChem CID
|
|
CompTox Dashboard (EPA)
|
|
| |
| |
| Properties | |
| B3H3O3 | |
| Molar mass | 83.455 g mol−1 |
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).
Infobox references
| |
Structure and bonding edit
Three-coordinate compounds of boron typically exhibit trigonal planar geometry, therefore the boroxine ring is locked in a planar geometry as well.[2][5] These compounds are isoelectronic to benzene. With the vacant p-orbital on the boron atoms, they may possess some aromatic character.[2][6] Boron single-bonds on boroxine compounds are mostly s-character.[5] Ethyl-substituted boroxine has B-O bond lengths of 1.384 Å and B-C bond lengths of 1.565 Å.[6] Phenyl-substituted boroxine has similar bond lengths of 1.386 Å and 1.546 Å respectively, showing that the substituent has little effect on the boroxine ring size.[6]
Substitutions onto a boroxine ring determine its crystal structure. Alkyl-substituted boroxines have the simplest crystal structure. These molecules stack on top of each other, aligning an oxygen atom from one molecule with a boron atom in another, leaving each boron atom between two other oxygen atoms. This forms a tube out of the individual boroxine rings. The intermolecular B-O distance of ethyl-substituted boroxine is 3.462 Å, which is much longer than the B-O bond distance of 1.384 Å. The crystal structure of phenyl-substituted boroxine is more complex. The interaction between the vacant p-orbitals in the boron atoms and the π-electrons in the aromatic, phenyl-substituents cause a different crystal structure. The boroxine ring of one molecule is stacked between two phenyl rings of other molecules. This arrangement allows the phenyl-substituents to donate π-electron density to the vacant boron p-orbitals.[6]
Synthesis edit
The parent boroxine (cyclo-(HBO)3) is prepared in small quantities as a low pressure gas by high temperature reaction of water and elemental boron or reaction of various boranes (B2H6 or B5H9) with O2. It is thermodynamically unstable with respect to disproportionation to diborane and boron oxide.[7] Some reactivity studies and an IR spectrum are reported, but it is otherwise not well characterized.[8]
As discovered in the 1930s, substituted boroxines (cyclo-(RBO)3, R = alkyl or aryl) are generally produced from their corresponding boronic acids by dehydration.[1][2][3] This dehydration can be done either by a drying agent or by heating under a high vacuum.[2]
Trimethylboroxine can be synthesized by reacting carbon monoxide with diborane (B2H6) and lithium borohydride (LiBH4) as a catalyst (or reaction of borane–tetrahydrofuran or borane–(dimethyl sulfide) in the presence of sodium borohydride):[5][9]
![{\displaystyle 3\ {\ce {CO}}+{\frac {3}{2}}\ {\ce {B2H6}}{\ce {->[{\ce {LiBH4 (catalyst)}}] (H3C BO)3}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/0d61e082719f2669e188673799d48b8693ed72a8)
Reactions edit
Trimethylboroxine is used in the methylation of various aryl halides through palladium-catalyzed Suzuki-Miyaura coupling reactions:[10]
![{\displaystyle {\ce {{\overset {(X = Br, I)}{C6H5X}}+ (CH3BO)3 ->[{\ce {K2CO3, Pd(PPh3)4}}][{\ce {dioxane}}] C6H5CH3}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/e07d87468caf83dee3007f299bb75ed8928deb4b)
Another form of the Suzuki-Miyaura coupling reaction exhibits selectivity to aryl chlorides:[11]
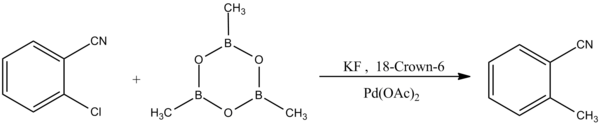
Reaction of boroxine

Boroxines have also been examined as precursors to monomeric oxoborane, HB≡O.[3] This compound quickly converts back to the cyclic boroxine, even at low temperatures.[3]
References edit
- ^ a b c Brown, H.C. Boranes in Organoc Chemistry; Cornell University Press: Ithaca, 1972; pp. 346–347.
- ^ a b c d e Hall, Dennis G. (2005). Boronic Acids – Preparation and Applications in Organic Synthesis and Medicine. John Wiley & Sons ISBN 3-527-30991-8.
- ^ a b c d Westcott, S.A. (2010). "BO Chemistry Comes Full Circle". Angewandte Chemie International Edition. 49 (48): 9045–9046. doi:10.1002/anie.201003379. PMID 20878818.
- ^ Wu, Q.G.; G. Wu; L. Leon Branca; S. Wang (1999). "B3O3Ph3 (7-Azaindole): Structure, Luminescence, and Fluxionality". Organometallics. 18 (13): 2552–2556. doi:10.1021/om990053t.
- ^ a b c Onak, T. in Organoborane Chemistry; Maitles, P.M., Stone, F.G.A., West, R., Eds.; Academic Press: New York, 1975; pp. 2,4,16,44.
- ^ a b c d Haberecht, M.C.; Bolte, Michael; Wagner, Matthias; Lerner, Hans-Wolfram (2005). "A New Polymorph of Tri(p-tolyl)boroxine". Journal of Chemical Crystallography. 35 (9): 657–665. doi:10.1007/s10870-005-3325-y.
- ^ Barton, L.; Grimm, F. A.; Porter, Richard F. (1966). "Boroxine: A Simplified Preparation". Inorganic Chemistry. 5 (11): 2076–2078. doi:10.1021/ic50045a062. ISSN 0020-1669.
- ^ Lee, George H.; Porter, Richard F. (1966). "Gaseous Boroxine: Mechanisms of Reactions with Oxygen and Carbon Monoxide". Inorganic Chemistry. 5 (8): 1329–1333. doi:10.1021/ic50042a007. ISSN 0020-1669.
- ^ Rathke, Michael W.; Brown, Herbert C. (1966). "A New Reaction of Diborane with Carbon Monoxide Catalyzed by Sodium Borohydride. A Convenient Synthesis of Trimethylboroxide". Journal of the American Chemical Society. 88 (11): 2606–2607. doi:10.1021/ja00963a054. ISSN 0002-7863.
- ^ Gray, M.; Andrews, I.P.; Hook, D.F.; Kitteringham, J.; Voyle, M. (2000). "Practical Methylation of Aryl Halides by Suzuki-Miyaura Coupling". Tetrahedron. 41 (32): 6237–6240. doi:10.1016/S0040-4039(00)01038-8.
- ^ Song, C.; Ma, Y.; Chai, Q.; Ma, C.; Jaing, W.; Andrus, M.B. (2005). "Palladium Catalyzed Suzuki-Miyaura Coupling With Aryl Chlorides Using a Bulky Phenanthryl N-heterocyclic Carbene Ligand". Tetrahedron. 61 (31): 7438–7446. doi:10.1016/j.tet.2005.05.071.