


Summary
Desulfonylation reactions are chemical reactions leading to the removal of a sulfonyl group from organic compounds. As the sulfonyl functional group is electron-withdrawing,[1] methods for cleaving the sulfur–carbon bonds of sulfones are typically reductive in nature. Olefination or replacement with hydrogen may be accomplished using reductive desulfonylation methods.[2]
Introduction edit
The sulfonyl functional group (RS(O)2R') has become an important electron-withdrawing group for modern organic chemistry. α-Sulfonyl carbanions may be used as nucleophiles in alkylation reactions, Michael-type additions, and other processes.[3] After having served their synthetic purpose, sulfonyl groups are often removed. In the presence of certain reducing agents, one of the sulfur-carbon bonds of the sulfonyl group is cleaved, leading to sulfur-free organic products. Depending on the nature of the substrate and reaction conditions, alkyl sulfones afford either the corresponding alkanes or olefins (the Julia olefination). Reductive desulfonylation is typically accomplished with active metals or salts (sodium amalgam, aluminium amalgam, magnesium, samarium(II) iodide), tin hydrides (tributyltin hydride), or transition metal complexes with reducing agents or nucleophiles (PdCl2(dppp)/LiHBEt3, Pd(PPh3)4/LiHBEt3, Pd(PPh3)4/NaHC(CO2Et)2). Alkyl, alkenyl, and allylic sulfones may be reduced using one or more of these methods.
(1)

Mechanism and stereochemistry edit
Reductive desulfonylation edit
Reductive desulfonylation reactions lead to the replacement of a carbon-sulfur bond in the sulfonyl group with a carbon-hydrogen bond. Because the sulfonyl group is by definition attached to two carbons, however, reduction to two sets of products is possible. Mechanistic studies of reductions employing metal amalgams as the reducing agent suggest that upon electron transfer to the sulfone, fragmentation to a sulfinate anion and the more stable organic radical occurs. Immediate reduction of the radical and protonation then occur to afford the sulfur-free product derived from the more stable radical. Thus, S-alkyl bonds are cleaved in preference to S-aryl or S-alkenyl bonds.[4]
-
(2)
![{\displaystyle {\begin{aligned}{\ce {ArSO2R->[+{\ce {e^{-}}}]{[ArSO2R]^{.-}}->{ArSO2^{-}}+}}\ &{\ce {R^{.}}}\\&{\ce {R^{.}->[+{\ce {e^{-}}}][{\ce {H-solv}}]R-H}}\\\end{aligned}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/af990b4b965c4b6ecf4e38875554dc167745a312)
Samarium(II) iodide may be used to reductively cleave α-keto sulfones;[5] in the presence of hexamethylphosphoramide (HMPA), SmI2 is able to effect reductive elimination of α-functionalized sulfones (see equation (11) below).
-
 (3)
(3)

Tin hydrides reduce α-keto[6] and allylic[7] sulfones. The mechanisms of these processes involve the addition of a tin-centered radical to the substrate followed by elimination of a sulfinyl radical, which abstracts a hydrogen from a molecule of tin hydride to propagate the radical chain. Protonation of the organotin intermediates thus formed (by sulfinic acid generated in situ) leads to reduced products. Addition of a stoichiometric amount of proton source allows the use of tin hydride in catalytic amounts. Although desulfonylations of allylic sulfones are site selective (providing only products of allylic transposition), they are not stereoselective and afford mixtures of double bond isomers.[7] The mechanism of desulfonylation of α-keto sulfones is similar.[6]
-
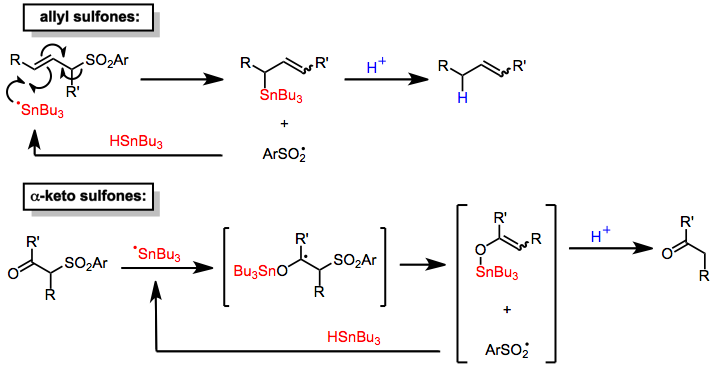 (4)
(4)

Transition-metal-mediated reductive desulfonylations rely on the generation of an intermediate π-allyl complex, which undergoes nucleophilic attack by hydride or another nucleophile to afford reduced products.[8] Nucleophilic attack generally occurs at the less substituted position of the π-allyl moiety, although site selectivity depends strongly on the substrate and reaction conditions. Palladium(0) complexes are the most commonly used precatalysts.
-
 (5)
(5)

Reductive elimination edit
Sulfones with a good leaving group in the β position may undergo reductive elimination under desulfonylation conditions to afford alkenes. This process is a key step of the Julia olefination, which yields alkenes via addition of an α-sulfonyl carbanion to an aldehyde followed by reductive elimination. Sodium amalgam[9] or samarium(II) iodide/HMPA[10] may be used to convert β-sulfonyloxy or β-acyloxy sulfones to the corresponding alkenes. The key mechanistic step of this process is elimination of an anionic or organometallic intermediate to generate the alkene.
(6)

The use of sodium amalgam, which promotes the formation of essentially free alkyl anions,[9] leads to (E) alkenes with extremely high selectivity. Samarium(II) iodide also produces the (E) alkene predominantly, but with lower selectivity.[10]
Scope and limitations edit
Using the appropriate reagent and conditions, alkyl, alkenyl, allylic, and α-keto sulfones may be reduced in good yield and high stereoselectivity (where applicable). Appropriate conditions for the reduction of these classes of sulfones are discussed below.
Alkyl sulfones may be reduced with sodium or lithium in liquid ammonia;[11] however, the strongly basic conditions of these dissolving metal reductions represent a significant disadvantage. In alcoholic solvents, magnesium metal and a catalytic amount of mercury(II) chloride may be used.[12] A wide variety of functional groups are unaffected by these conditions, including many that are transformed by dissolving metal reductions. Reductive desulfonylation with these reagents does not occur in reactions of β-hydroxy sulfones, due to the poor leaving group ability of the hydroxyl group.[13]
-
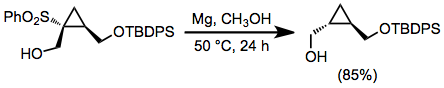 (7)
(7)
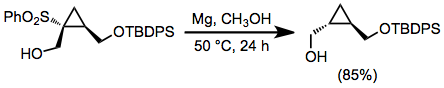
A significant issue associated with the reduction of allylic sulfones is transposition of the allylic double bond, which occurs in varying amounts during reductions by metal amalgams.[14] and tin hydrides[15] Palladium-catalyzed reductive desulfonylations of allylic sulfones do not have this issue, and afford allylic sulfones with high site and stereoselectivity.[8]
-
 (8)
(8)

Aluminum amalgam (Al/Hg) may be used for the chemoselective reduction of α-sulfonylated carbonyl groups. Carboxylic acid derivatives, acetals, thioacetals, amines, alcohols, and isolated double bonds are all inert to Al/Hg. Selective desulfonylation may be carried out on β-hydroxy sulfones without reductive elimination.[16]
-
 (9)
(9)

Transition metal catalysis is also useful for the stereospecific reduction of alkenyl sulfones. In the presence of an excess of a Grignard reagent, a palladium(II) or nickel(II) catalyst, and a phosphorus or nitrogen ligand, alkenyl sulfones are converted to the corresponding alkenes stereospecifically in good yield. On the other hand, dissolving metal and metal amalgam reductions are not stereoselective in general.[17] Palladium catalysis is generally superior to nickel catalysis, giving higher yields and stereoselectivities.[18][19]
-
 (10)
(10)

Alkyl and alkenyl sulfones with good leaving groups in the β position undergo elimination under reductive conditions to afford alkenes or alkynes. The Julia olefination exploits this process for the synthesis of alkenes from alkyl sulfones and carbonyl compounds. Addition of an α-sulfonyl anion to a carbonyl compound, followed by quenching with an acyl or sulfonyl chloride, leads to a β-acyloxy or -sulfonyloxy sulfone, which undergoes elimination under reductive conditions. Sodium amalgam may be used to accomplish the elimination step;[9] however, the combination of samarium(II) iodide and HMPA is milder than strongly basic sodium amalgam and leads to higher yields in reductive elimination processes.[20]
-
 (11)
(11)

Synthetic applications edit
The modest acidity of carbons adjacent to the sulfonyl group has made sulfones useful for organic synthesis. Upon removal of the sulfonyl group with desulfonylation or reductive elimination, the net result is the formation of a carbon-carbon bond single or double bond between two unfunctionalized carbons, a ubiquitous motif in synthetic targets. In a synthesis of (–)-anthoplalone, Julia olefination was used to establish the (E)-alkene in the target.[21]
(12)

Reductive desulfonylation is employed when the establishment of a carbon-carbon single bond is the goal. In a synthesis of (+)-chatancin, alkylation of an α-sulfonyl carbanion followed by desulfonylation established a key allylic carbon-carbon bond.[22]
(13)

Comparison with other methods edit
As α-cyano carbanions may be used in many of the same contexts as α-sulfonyl anions, reductive decyanation methods offer a viable alternative to reductive desulfonylation. Dissolving metal reductions are most useful for the decyanation of tertiary nitriles (primary and secondary nitriles give the corresponding amines in addition to decyanated products),[23] but potassium is a more general reducing agent that reduces primary, secondary, and tertiary nitriles.[24]
(14)

A wide variety of carbonyl olefination methods that are direct alternative to the Julia olefination are known: the Wittig reaction,[25] the Horner-Wadsworth-Emmons reaction,[26] Peterson olefination,[27] and others. The primary advantage of Julia olefination is that the sulfone precursors are sometimes more readily available and easier to purify than the corresponding phosphorus- or silicon-containing compounds. Additionally, a variety of methods to synthesize sulfones exist.[28] Nonetheless, the sometimes limited stereoselectivity (and in particular, the difficulty of accessing (Z) -alkenes) of the Julia reaction can be problematic. Many alternative methods for olefination, including the Peterson reaction,[27] do not have this issue.
(15)

References edit
- ^ Kharasch, Norman; Meyers, Cal Y. (2013-10-22). The Chemistry of Organic Sulfur Compounds. Elsevier. ISBN 978-1-4831-5611-8.
- ^ Alonso, Diego A.; Ájera, Carmen N (2009). "Desulfonylation Reactions". Organic Reactions. doi:10.1002/0471264180.or072.02. ISBN 978-0471264187.
- ^ Prilezhaeva, E. (2000). "Sulfones and sulfoxides in the total synthesis of biologically active natural compounds". Russ. Chem. Rev. 69 (5): 367–408. Bibcode:2000RuCRv..69..367P. doi:10.1070/RC2000v069n05ABEH000561. S2CID 250835581.
- ^ Horner, L.; Neumann, H. (1965). "Studien zum Vorgang der Wasserstoffübertragung, XII: Hydrierende Spaltung von Sulfonen mit Tetramethylammonium als Elektronenüberträger". Chem. Ber. 98 (6): 1715. doi:10.1002/cber.19650980606.
- ^ Molander, Gary A. (1994-10-27), John Wiley & Sons, Inc. (ed.), "Reductions with Samarium(II) Iodide", Organic Reactions, Hoboken, NJ, USA: John Wiley & Sons, Inc., pp. 211–367, doi:10.1002/0471264180.or046.03, ISBN 978-0-471-26418-7, retrieved 2022-02-27
- ^ a b Wnuk, Stanislaw F.; Rios, Jeannette M.; Khan, Jahanzeb; Hsu, Ya-Li (2000). "Stannyl Radical-Mediated Cleavage of π-Deficient Heterocyclic Sulfones. Synthesis of α-Fluoro Esters". The Journal of Organic Chemistry. 65 (13): 4169–74. doi:10.1021/jo000342n. PMID 10866636. S2CID 94913752.
- ^ a b Ueno, Y.; Aoki, S.; Okawara, M. (1979). "Synthetic reactions using organotin and sulfur compounds. 3. Regioselective desulfonylation of allylic sulfones with organotin hydride involving double migration of the double bond". Journal of the American Chemical Society. 101 (18): 5414. doi:10.1021/ja00512a051.
- ^ a b Hutchins, Robert O.; Learn, Keith (1982). "Regio- and stereoselective reductive replacement of allylic oxygen, sulfur, and selenium functional groups by hydride via catalytic activation by palladium(0) complexes". The Journal of Organic Chemistry. 47 (22): 4380. doi:10.1021/jo00143a054.
- ^ a b c Kocienski, Philip J.; Lythgoe, Basil; Waterhouse, Ian (1980). "The influence of chain-branching on the steric outcome of some olefin-forming reactions". Journal of the Chemical Society, Perkin Transactions 1: 1045. doi:10.1039/P19800001045.
- ^ a b Markó, I; Murphy, Fiona; Dolan, Simon (1996). "Efficient preparation of trisubstituted alkenes using the Julia-Lythgoe olefination of ketones. On the key-role of SmI2 in the reductive elimination step". Tetrahedron Letters. 37 (12): 2089. doi:10.1016/0040-4039(96)00200-6.
- ^ Sato, Kikumasa; Inoue, Seiichi; Onishi, Akira; Uchida, Nobuhiko; Minowa, Nobuto (1981). "Stereoselective synthesis of solanesol and all-trans-decaprenol". Journal of the Chemical Society, Perkin Transactions 1: 761. doi:10.1039/P19810000761.
- ^ Lai, J; Yu, Jurong; David Hawkins, R.; Falck, J.R. (1995). "Two-carbon elongation/annulation of alcohols to nitriles". Tetrahedron Letters. 36 (32): 5691. doi:10.1016/0040-4039(95)01125-2.
- ^ Kazuta, Yuji; Matsuda, Akira; Shuto, Satoshi (2002). "Development of Versatile cis- and trans-Dicarbon-Substituted Chiral Cyclopropane Units: Synthesis of (1S,2R)- and (1R,2R)-2-Aminomethyl-1-(1H-imidazol-4-yl)cyclopropanes and Their Enantiomers as Conformationally Restricted Analogues of Histamine". The Journal of Organic Chemistry. 67 (5): 1669–77. doi:10.1021/jo010852x. PMID 11871901.
- ^ Alonso, Diego A.; Falvello, Larry R.; Mancheño, Balbino; Nájera, Carmen; Tomás, Milagros (1996). "Lithiated γ-Tosyl-Substituted Benzylmethallylamine: New γ-Amino Methallyl Sulfone Anions in Organic Synthesis†". The Journal of Organic Chemistry. 61 (15): 5004. doi:10.1021/jo9602478.
- ^ Ueno, Y; Sano, Hiroshi; Aoki, Seiichi; Okawara, Makoto (1981). "Stannes in synthesis: A new route to 2-substituted-1,3-butadienes via stereoselective allyltin formation under homolytic conditions". Tetrahedron Letters. 22 (28): 2675. doi:10.1016/S0040-4039(01)92967-3.
- ^ Nanda, Samik (2005). "Chemoenzymatic total synthesis of the phytotoxic lactone herbarumin III". Tetrahedron Letters. 46 (21): 3661–3663. doi:10.1016/j.tetlet.2005.03.139.
- ^ Caturla, F; Nájera, Carmen (1997). "Preparation and synthetic applications of lithiated vinyl sulfones derived from 3-buten-1-ol and 4-penten-1-ol". Tetrahedron. 53 (33): 11449. doi:10.1016/S0040-4020(97)00725-4.
- ^ Fabre, J; Julia, M (1983). "Organic Synthesis with Sulfones noXXIX Stereospecific hydrogenolysis of vinylic sulfones with grignards and transition metal catalysts". Tetrahedron Letters. 24 (40): 4311. doi:10.1016/S0040-4039(00)88328-8.
- ^ Cuvigny, T; Du Penhoat, C.Herve; Julia, M. (1987). "Syntheses with sulfones XLVII : stereoselective access to 1,3- and 1,4-dienes through hydrogenolysis of benzenesulfonyldienes. Application to pheromone synthesis". Tetrahedron. 43 (5): 859. doi:10.1016/S0040-4020(01)90023-7.
- ^ Ihara, M.; Suzuki, S.; Taniguchi, T.; Tokunaga, Y.; Fukumoto, K. (1994). "Modification of the Julia Alkenation Using SmI2-HMPA". Synlett. 1994 (10): 859. doi:10.1055/s-1994-23033.
- ^ Hanessian, Stephen; Cantin, Louis-David; Andreotti, Daniele (1999). "Total Synthesis and Absolute Configuration of (−)-Anthoplalone". The Journal of Organic Chemistry. 64 (13): 4893–4900. doi:10.1021/jo990302n. PMID 11674567.
- ^ Shindo, Mitsuru; Sugioka, Tomoyuki; Umaba, Yuko; Shishido, Kozo (2004). "Total synthesis of (+)-bongkrekic acid". Tetrahedron Letters. 45 (48): 8863. doi:10.1016/j.tetlet.2004.09.162.
- ^ Arapakos, P. G.; Scott, Malcolm K.; Huber, F. E. (1969). "Reaction of nitriles with solvated electrons. III". Journal of the American Chemical Society. 91 (8): 2059. doi:10.1021/ja01036a033.
- ^ Wender, a; Delong, Mitch A. (1990). "Synthetic studies on arene-olefin cycloadditions. XII. Total synthesis of (±)-subergorgic acid". Tetrahedron Letters. 31 (38): 5429. doi:10.1016/S0040-4039(00)97864-X.
- ^ Vedejs, E.; Peterson, M. J. (1994). "Stereochemistry and Mechanism in the Wittig Reaction". Top. Stereochem. Topics in Stereochemistry. 21: 1–157. doi:10.1002/9780470147306.ch1. ISBN 9780470147306.
- ^ Wadsworth, W. S. (1977). "Synthetic Applications of Phosphoryl-Stabilized Anions". Org. React. 25: 73–253. doi:10.1002/0471264180.or025.02. ISBN 0471264180.
- ^ a b Peterson, Donald John (1968). "Carbonyl olefination reaction using silyl-substituted organometallic compounds". The Journal of Organic Chemistry. 33 (2): 780–784. doi:10.1021/jo01266a061.
- ^ Simpkins, N. S. Sulphones in Organic Synthesis; Pergamon Press: Oxford, 1993.