


Summary
Fluorescence correlation spectroscopy (FCS) is a statistical analysis, via time correlation, of stationary fluctuations of the fluorescence intensity. Its theoretical underpinning originated from L. Onsager's regression hypothesis. The analysis provides kinetic parameters of the physical processes underlying the fluctuations. One of the interesting applications of this is an analysis of the concentration fluctuations of fluorescent particles (molecules) in solution. In this application, the fluorescence emitted from a very tiny space in solution containing a small number of fluorescent particles (molecules) is observed. The fluorescence intensity is fluctuating due to Brownian motion of the particles. In other words, the number of the particles in the sub-space defined by the optical system is randomly changing around the average number. The analysis gives the average number of fluorescent particles and average diffusion time, when the particle is passing through the space. Eventually, both the concentration and size of the particle (molecule) are determined. Both parameters are important in biochemical research, biophysics, and chemistry.
FCS is such a sensitive analytical tool because it observes a small number of molecules (nanomolar to picomolar concentrations) in a small volume (~1 μm3).[1] In contrast to other methods (such as HPLC analysis) FCS has no physical separation process; instead, it achieves its spatial resolution through its optics. Furthermore, FCS enables observation of fluorescence-tagged molecules in the biochemical pathway in intact living cells.[2] This opens a new area, "in situ or in vivo biochemistry": tracing the biochemical pathway in intact cells and organs.[3]
Commonly, FCS is employed in the context of optical microscopy, in particular confocal microscopy or two-photon excitation microscopy. In these techniques light is focused on a sample and the measured fluorescence intensity fluctuations (due to diffusion, physical or chemical reactions, aggregation, etc.) are analyzed using the temporal autocorrelation. Because the measured property is essentially related to the magnitude and/or the amount of fluctuations, there is an optimum measurement regime at the level when individual species enter or exit the observation volume (or turn on and off in the volume). When too many entities are measured at the same time the overall fluctuations are small in comparison to the total signal and may not be resolvable – in the other direction, if the individual fluctuation-events are too sparse in time, one measurement may take prohibitively too long. FCS is in a way the fluorescent counterpart to dynamic light scattering, which uses coherent light scattering, instead of (incoherent) fluorescence.
When an appropriate model is known, FCS can be used to obtain quantitative information such as
- diffusion coefficients
- hydrodynamic radii
- average concentrations
- kinetic chemical reaction rates
- singlet-triplet dynamics
Because fluorescent markers come in a variety of colors and can be specifically bound to a particular molecule (e.g. proteins, polymers, metal-complexes, etc.), it is possible to study the behavior of individual molecules (in rapid succession in composite solutions). With the development of sensitive detectors such as avalanche photodiodes the detection of the fluorescence signal coming from individual molecules in highly dilute samples has become practical. With this emerged the possibility to conduct FCS experiments in a wide variety of specimens, ranging from materials science to biology. The advent of engineered cells with genetically tagged proteins (like green fluorescent protein) has made FCS a common tool for studying molecular dynamics in living cells.[4]
History edit
Signal-correlation techniques were first experimentally applied to fluorescence in 1972 by Magde, Elson, and Webb,[5] who are therefore commonly credited as the "inventors" of FCS. The technique was further developed in a group of papers by these and other authors soon after, establishing the theoretical foundations and types of applications.[6][7][8] Around 1990, with the ability of detecting sufficiently small number of fluorescence particles, two issues emerged: A non-Gaussian distribution of the fluorescence intensity and the three-dimensional confocal Measurement Volume of a laser-microscopy system.[9] The former led to an analysis of distributions and moments of the fluorescent signals for extracting molecular information,[10][11] which eventually became a collection of methods known as Brightness Analyses. See Thompson (1991)[12] for a review of that period.
Beginning in 1993,[13] a number of improvements in the measurement techniques—notably using confocal microscopy, and then two-photon microscopy—to better define the measurement volume and reject background—greatly improved the signal-to-noise ratio and allowed single molecule sensitivity.[14][15] Since then, there has been a renewed interest in FCS, and as of August 2007 there have been over 3,000 papers using FCS found in Web of Science. See Krichevsky and Bonnet[16] for a review. In addition, there has been a flurry of activity extending FCS in various ways, for instance to laser scanning and spinning-disk confocal microscopy (from a stationary, single point measurement), in using cross-correlation (FCCS) between two fluorescent channels instead of autocorrelation, and in using Förster Resonance Energy Transfer (FRET) instead of fluorescence.
Typical setup edit

The typical FCS setup consists of a laser line (wavelengths ranging typically from 405–633 nm (cw), and from 690–1100 nm (pulsed)), which is reflected into a microscope objective by a dichroic mirror. The laser beam is focused in the sample, which contains fluorescent particles (molecules) in such high dilution, that only a few are within the focal spot (usually 1–100 molecules in one fL). When the particles cross the focal volume, they fluoresce. This light is collected by the same objective and, because it is red-shifted with respect to the excitation light it passes the dichroic mirror reaching a detector, typically a photomultiplier tube, an avalanche photodiode detector or a superconducting nanowire single-photon detector. The resulting electronic signal can be stored either directly as an intensity versus time trace to be analyzed at a later point, or computed to generate the autocorrelation directly (which requires special acquisition cards). The FCS curve by itself only represents a time-spectrum. Conclusions on physical phenomena have to be extracted from there with appropriate models. The parameters of interest are found after fitting the autocorrelation curve to modeled functional forms.[17]
Measurement volume edit
The measurement volume is a convolution of illumination (excitation) and detection geometries, which result from the optical elements involved. The resulting volume is described mathematically by the point spread function (or PSF), it is essentially the image of a point source. The PSF is often described as an ellipsoid (with unsharp boundaries) of few hundred nanometers in focus diameter, and almost one micrometer along the optical axis. The shape varies significantly (and has a large impact on the resulting FCS curves) depending on the quality of the optical elements (it is crucial to avoid astigmatism and to check the real shape of the PSF on the instrument). In the case of confocal microscopy, and for small pinholes (around one Airy unit), the PSF is well approximated by Gaussians:

where is the peak intensity, r and z are radial and axial position, and and are the radial and axial radii, and . This Gaussian form is assumed in deriving the functional form of the autocorrelation.




Typically is 200–300 nm, and is 2–6 times larger.[18] One common way of calibrating the measurement volume parameters is to perform FCS on a species with known diffusion coefficient and concentration (see below). Diffusion coefficients for common fluorophores in water are given in a later section.
The Gaussian approximation works to varying degrees depending on the optical details, and corrections can sometimes be applied to offset the errors in approximation.[19]
Autocorrelation function edit

The (temporal) autocorrelation function is the correlation of a time series with itself shifted by time , as a function of :


where is the deviation from the mean intensity. The normalization (denominator) here is the most commonly used for FCS, because then the correlation at , G(0), is related to the average number of particles in the measurement volume.


As an example, raw FCS data and its autocorrelation for freely diffusing Rhodamine 6G are shown in the figure to the right. The plot on top shows the fluorescent intensity versus time. The intensity fluctuates as Rhodamine 6G moves in and out of the focal volume. In the bottom plot is the autocorrelation on the same data. Information about the diffusion rate and concentration can be obtained using one of the models described below.
For a Gaussian illumination profile , the autocorrelation function is given by the general master formula[20]


where the vector denotes the stochastic displacement in space of a fluorophore after time . The expression is valid if the average number of fluorophores in the focal volume is low and if dark states, etc., of the fluorophore can be ignored. In particular, no assumption was made on the type of diffusive motion under investigation. The formula allows for an interpretation of as (i) a return probability for small beam parameters and (ii) the moment-generating function of if are varied.





Interpreting the autocorrelation function edit
To extract quantities of interest, the autocorrelation data can be fitted, typically using a nonlinear least squares algorithm. The fit's functional form depends on the type of dynamics (and the optical geometry in question).
Normal diffusion edit
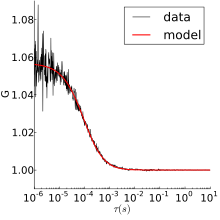
The fluorescent particles used in FCS are small and thus experience thermal motions in solution. The simplest FCS experiment is thus normal 3D diffusion, for which the autocorrelation is:

where is the ratio of axial to radial radii of the measurement volume, and is the characteristic residence time. This form was derived assuming a Gaussian measurement volume. Typically, the fit would have three free parameters—G(0), , and —from which the diffusion coefficient and fluorophore concentration can be obtained.




With the normalization used in the previous section, G(0) gives the mean number of diffusers in the volume <N>, or equivalently—with knowledge of the observation volume size—the mean concentration:

where the effective volume is found from integrating the Gaussian form of the measurement volume and is given by:

- D gives the diffusion coefficient:

Anomalous diffusion edit
If the diffusing particles are hindered by obstacles or pushed by a force (molecular motors, flow, etc.) the dynamics is often not sufficiently well-described by the normal diffusion model, where the mean squared displacement (MSD) grows linearly with time. Instead the diffusion may be better described as anomalous diffusion, where the temporal dependence of the MSD is non-linear as in the power-law:

where is an anomalous diffusion coefficient. "Anomalous diffusion" commonly refers only to this very generic model, and not the many other possibilities that might be described as anomalous. Also, a power law is, in a strict sense, the expected form only for a narrow range of rigorously defined systems, for instance when the distribution of obstacles is fractal. Nonetheless a power law can be a useful approximation for a wider range of systems.

The FCS autocorrelation function for anomalous diffusion is:

where the anomalous exponent is the same as above, and becomes a free parameter in the fitting.

Using FCS, the anomalous exponent has been shown to be an indication of the degree of molecular crowding (it is less than one and smaller for greater degrees of crowding).[21]
Polydisperse diffusion edit
If there are diffusing particles with different sizes (diffusion coefficients), it is common to fit to a function that is the sum of single component forms:

where the sum is over the number different sizes of particle, indexed by i, and gives the weighting, which is related to the quantum yield and concentration of each type. This introduces new parameters, which makes the fitting more difficult as a higher-dimensional space must be searched. Nonlinear least square fitting typically becomes unstable with even a small number of s. A more robust fitting scheme, especially useful for polydisperse samples, is the Maximum Entropy Method.[22]


Diffusion with flow edit
With diffusion together with a uniform flow with velocity in the lateral direction, the autocorrelation is:[23]

![{\displaystyle \ G(\tau )=G(0){\frac {1}{(1+(\tau /\tau _{D}))(1+a^{-2}(\tau /\tau _{D}))^{1/2}}}\times \exp[-(\tau /\tau _{v})^{2}\times {\frac {1}{1+\tau /\tau _{D}}}]+G(\infty )}](https://wikimedia.org/api/rest_v1/media/math/render/svg/2338429881fea18317141e28d9c1accc4c59f52c)
where is the average residence time if there is only a flow (no diffusion).

Chemical relaxation edit
A wide range of possible FCS experiments involve chemical reactions that continually fluctuate from equilibrium because of thermal motions (and then "relax"). In contrast to diffusion, which is also a relaxation process, the fluctuations cause changes between states of different energies. One very simple system showing chemical relaxation would be a stationary binding site in the measurement volume, where particles only produce signal when bound (e.g. by FRET, or if the diffusion time is much faster than the sampling interval). In this case the autocorrelation is:

where

is the relaxation time and depends on the reaction kinetics (on and off rates), and:

is related to the equilibrium constant K.
Most systems with chemical relaxation also show measurable diffusion as well, and the autocorrelation function will depend on the details of the system. If the diffusion and chemical reaction are decoupled, the combined autocorrelation is the product of the chemical and diffusive autocorrelations.
Triplet state correction edit
The autocorrelations above assume that the fluctuations are not due to changes in the fluorescent properties of the particles. However, for the majority of (bio)organic fluorophores—e.g. green fluorescent protein, rhodamine, Cy3 and Alexa Fluor dyes—some fraction of illuminated particles are excited to a triplet state (or other non-radiative decaying states) and then do not emit photons for a characteristic relaxation time . Typically is on the order of microseconds, which is usually smaller than the dynamics of interest (e.g. ) but large enough to be measured. A multiplicative term is added to the autocorrelation to account for the triplet state. For normal diffusion:


where is the fraction of particles that have entered the triplet state and is the corresponding triplet state relaxation time. If the dynamics of interest are much slower than the triplet state relaxation, the short time component of the autocorrelation can simply be truncated and the triplet term is unnecessary.


Common fluorescent probes edit
The fluorescent species used in FCS is typically a biomolecule of interest that has been tagged with a fluorophore (using immunohistochemistry for instance), or is a naked fluorophore that is used to probe some environment of interest (e.g. the cytoskeleton of a cell). The following table gives diffusion coefficients of some common fluorophores in water at room temperature, and their excitation wavelengths.
| Fluorescent dye | [10−10 m2 s−1] |
T [°C] | Excitation wavelength [nm] |
Reference |
|---|---|---|---|---|
| Rhodamine 6G | 2.8, 3.0, 4.14 ± 0.05, 4.20 ± 0.06 | 25 | 514 | [8][25][26][27] |
| Rhodamine 110 | 2.7 | 488 | [28] | |
| Tetramethyl rhodamine | 2.6 | 543 | ||
| Cy3 | 2.8 | 543 | ||
| Cy5 | 2.5, 3.7 ± 0.15 | 25 | 633 | [29][30] |
| carboxyfluorescein | 3.2 | 488 | ||
| Alexa 488 | 1.96, 4.35 | 22.5±0.5 | 488 | [28][31] |
| Atto 655-maleimide | 4.07 ± 0.1 | 25 | 663 | [26] |
| Atto 655-carboxylicacid | 4.26 ± 0.08 | 25 | 663 | [26] |
| 2′, 7′-difluorofluorescein (Oregon Green 488) |
4.11 ± 0.06 | 25 | 498 | [26] |

Variations edit
FCS almost always refers to the single point, single channel, temporal autocorrelation measurement, although the term "fluorescence correlation spectroscopy" out of its historical scientific context implies no such restriction. FCS has been extended in a number of variations by different researchers, with each extension generating another name (usually an acronym).
Spot variation fluorescence correlation spectroscopy (svFCS) edit
Whereas FCS is a point measurement providing diffusion time at a given observation volume, svFCS is a technique where the observation spot is varied in order to measure diffusion times at different spot sizes. The relationship between the diffusion time and the spot area is linear and could be plotted in order to decipher the major contribution of confinement. The resulting curve is called the diffusion law. This technique is used in Biology to study the plasma membrane organization on living cells.

where is the y axis intercept. In case of Brownian diffusion, . In case of a confinement due to isolated domains, whereas in case of isolated domains, .




svFCS studies on living cells and simulation papers[32][33][34][35][36]
Sampling-Volume-Controlled Fluorescence Correlation Spectroscopy (SVC-FCS):[37]
z-scan FCS[38]
FCS with Nano-apertures: breaking the diffraction barrier[39]
STED-FCS:[40]
Fluorescence cross-correlation spectroscopy (FCCS) edit
FCS is sometimes used to study molecular interactions using differences in diffusion times (e.g. the product of an association reaction will be larger and thus have larger diffusion times than the reactants individually); however, FCS is relatively insensitive to molecular mass as can be seen from the following equation relating molecular mass to the diffusion time of globular particles (e.g. proteins):

where is the viscosity of the sample and is the molecular mass of the fluorescent species. In practice, the diffusion times need to be sufficiently different—a factor of at least 1.6—which means the molecular masses must differ by a factor of 4.[41] Dual color fluorescence cross-correlation spectroscopy (FCCS) measures interactions by cross-correlating two or more fluorescent channels (one channel for each reactant), which distinguishes interactions more sensitively than FCS, particularly when the mass change in the reaction is small.


Brightness analysis methods edit
This set of methods include number and brightness (N&B),[42] photon counting histogram (PCH),[43] fluorescence intensity distribution analysis (FIDA),[44] and Cumulant Analysis.[45] and Spatial Intensity Distribution Analysis.[46] Combination of multiple methods is also reported.[47] Fluorescence cross correlation spectroscopy overcomes the weak dependence of diffusion rate on molecular mass by looking at multicolor coincidence. What about homo-interactions? The solution lies in brightness analysis. These methods use the heterogeneity in the intensity distribution of fluorescence to measure the molecular brightness of different species in a sample. Since dimers will contain twice the number of fluorescent labels as monomers, their molecular brightness will be approximately double that of monomers. As a result, the relative brightness is sensitive a measure of oligomerization. The average molecular brightness ( ) is related to the variance ( ) and the average intensity ( ) as follows:[48]




Here and are the fractional intensity and molecular brightness, respectively, of species .



FRET-FCS edit
Another FCS based approach to studying molecular interactions uses fluorescence resonance energy transfer (FRET) instead of fluorescence, and is called FRET-FCS.[49] With FRET, there are two types of probes, as with FCCS; however, there is only one channel and light is only detected when the two probes are very close—close enough to ensure an interaction. The FRET signal is weaker than with fluorescence, but has the advantage that there is only signal during a reaction (aside from autofluorescence).
Scanning FCS edit
In Scanning fluorescence correlation spectroscopy (sFCS) the measurement volume is moved across the sample in a defined way. The introduction of scanning is motivated by its ability to alleviate or remove several distinct problems often encountered in standard FCS, and thus, to extend the range of applicability of fluorescence correlation methods in biological systems.[50]
Some variations of FCS are only applicable to serial scanning laser microscopes. Image Correlation Spectroscopy and its variations all were implemented on a scanning confocal or scanning two photon microscope, but transfer to other microscopes, like a spinning disk confocal microscope. Raster ICS (RICS),[51] and position sensitive FCS (PSFCS)[52] incorporate the time delay between parts of the image scan into the analysis. Also, low-dimensional scans (e.g. a circular ring)[53]—only possible on a scanning system—can access time scales between single point and full image measurements. Scanning path has also been made to adaptively follow particles.[54]
Spinning disk FCS and spatial mapping edit
Any of the image correlation spectroscopy methods can also be performed on a spinning disk confocal microscope, which in practice can obtain faster imaging speeds compared to a laser scanning confocal microscope. This approach has recently been applied to diffusion in a spatially varying complex environment, producing a pixel resolution map of a diffusion coefficient.[55] The spatial mapping of diffusion with FCS has subsequently been extended to the TIRF system.[56] Spatial mapping of dynamics using correlation techniques had been applied before, but only at sparse points[57] or at coarse resolution.[58]
Image correlation spectroscopy (ICS) edit
When the motion is slow (in biology, for example, diffusion in a membrane), getting adequate statistics from a single-point FCS experiment may take a prohibitively long time. More data can be obtained by performing the experiment in multiple spatial points in parallel, using a laser scanning confocal microscope. This approach has been called Image Correlation Spectroscopy (ICS).[59] The measurements can then be averaged together.
Another variation of ICS performs a spatial autocorrelation on images, which gives information about the concentration of particles.[60] The correlation is then averaged in time. While camera white noise does not autocorrelate over time, it does over space - this creates a white noise amplitude in the spatial autocorrelation function which must be accounted for when fitting the autocorrelation amplitude in order to find the concentration of fluorescent molecules.
A natural extension of the temporal and spatial correlation versions is spatio-temporal ICS (STICS).[58] In STICS there is no explicit averaging in space or time (only the averaging inherent in correlation). In systems with non-isotropic motion (e.g. directed flow, asymmetric diffusion), STICS can extract the directional information. A variation that is closely related to STICS (by the Fourier transform) is k-space Image Correlation Spectroscopy (kICS).[61]
There are cross-correlation versions of ICS as well, which can yield the concentration, distribution and dynamics of co-localized fluorescent molecules.[59] Molecules are considered co-localized when individual fluorescence contributions are indistinguishable due to overlapping point-spread functions of fluorescence intensities.
Particle image correlation spectroscopy (PICS) edit
Source:[62]
PICS is a powerful analysis tool that resolves correlations on the nanometer length and millisecond timescale. Adapted from methods of spatio-temporal image correlation spectroscopy,[58] it exploits the high positional accuracy of single-particle tracking. While conventional tracking methods break down if multiple particle trajectories intersect, this method works in principle for arbitrarily large molecule densities and dynamical parameters (e.g. diffusion coefficients, velocities) as long as individual molecules can be identified. It is computationally cheap and robust and allows one to identify and quantify motions (e.g. diffusion, active transport, confined diffusion) within an ensemble of particles, without any a priori knowledge about the dynamics.
A particle image cross-correlation spectroscopy (PICCS) extension is available for biological processes that involve multiple interaction partners, as can observed by two-color microscopy.[63]
FCS Super-resolution Optical Fluctuation Imaging (fcsSOFI) edit
Super-resolution optical fluctuation imaging (SOFI) is a super-resolution technique that achieves spatial resolutions below the diffraction limit by post-processing analysis with correlation equations, similar to FCS. While original reports of SOFI used fluctuations from stationary, blinking of fluorophores, FCS has been combined with SOFI where fluctuations are produced from diffusing probes to produce super-resolution spatial maps of diffusion coefficients.[64] This has been applied to understand diffusion and spatial properties of porous and confined materials. This includes agarose[64] and temperature-responsive PNIPAM hydrogels,[65] liquid crystals,[64] and phase-separated polymers and RNA/protein condensates.[66]
Total internal reflection FCS edit
Total internal reflection fluorescence (TIRF) is a microscopy approach that is only sensitive to a thin layer near the surface of a coverslip, which greatly minimizes background fluorescence. FCS has been extended to that type of microscope, and is called TIR-FCS.[67] Because the fluorescence intensity in TIRF falls off exponentially with distance from the coverslip (instead of as a Gaussian with a confocal), the autocorrelation function is different.
FCS imaging using Light sheet fluorescence microscopy edit
Light sheet fluorescence microscopy or selective plane imaging microscopy (SPIM) uses illumination that is done perpendicularly to the direction of observation, by using a thin sheet of (laser) light. Under certain conditions, this illumination principle can be combined with fluorescence correlation spectroscopy, to allow spatially resolved imaging of the mobility and interactions of fluorescing particles such as GFP labelled proteins inside living biological samples.[68]
Other fluorescent dynamical approaches edit
There are two main non-correlation alternatives to FCS that are widely used to study the dynamics of fluorescent species.
Fluorescence recovery after photobleaching (FRAP) edit
In FRAP, a region is briefly exposed to intense light, irrecoverably photobleaching fluorophores, and the fluorescence recovery due to diffusion of nearby (non-bleached) fluorophores is imaged. A primary advantage of FRAP over FCS is the ease of interpreting qualitative experiments common in cell biology. Differences between cell lines, or regions of a cell, or before and after application of drug, can often be characterized by simple inspection of movies. FCS experiments require a level of processing and are more sensitive to potentially confounding influences like: rotational diffusion, vibrations, photobleaching, dependence on illumination and fluorescence color, inadequate statistics, etc. It is much easier to change the measurement volume in FRAP, which allows greater control. In practice, the volumes are typically larger than in FCS. While FRAP experiments are typically more qualitative, some researchers are studying FRAP quantitatively and including binding dynamics.[69] A disadvantage of FRAP in cell biology is the free radical perturbation of the cell caused by the photobleaching. It is also less versatile, as it cannot measure concentration or rotational diffusion, or co-localization. FRAP requires a significantly higher concentration of fluorophores than FCS.
Particle tracking edit
In particle tracking, the trajectories of a set of particles are measured, typically by applying particle tracking algorithms to movies.[1] Particle tracking has the advantage that all the dynamical information is maintained in the measurement, unlike FCS where correlation averages the dynamics to a single smooth curve. The advantage is apparent in systems showing complex diffusion, where directly computing the mean squared displacement allows straightforward comparison to normal or power law diffusion. To apply particle tracking, the particles have to be distinguishable and thus at lower concentration than required of FCS. Also, particle tracking is more sensitive to noise, which can sometimes affect the results unpredictably.
Auto-fluorescence correlation spectroscopy edit
Recent advances in ultraviolet nanophotonics has led to development of single molecule study on label-free protein by exciting them with deep ultraviolet light and studying the dynamic processes.[70] [71] [72]
Two- and three-photon FCS excitation edit
Several advantages in both spatial resolution and minimizing photodamage/photobleaching in organic and/or biological samples are obtained by two-photon or three-photon excitation FCS.[73][74][75][76][77]
See also edit
References edit
- ^ Chen, H., Farkas, E., & Webb, W. (2008). In vivo applications of fluorescence correlation spectroscopy. Biophysical Tools for Biologists, Vol 2: In Vivo Techniques, 89, 3-+.
- ^ Kwapiszewska, K.; Szczepański, K.; Kalwarczyk, T.; Michalska, B.; Patalas-Krawczyk, P.; Szymański, J.; Andryszewski, T.; Iwan, M.; Duszyński, J.; Hołyst, R. (2020). "Nanoscale Viscosity of Cytoplasm Is Conserved in Human Cell Lines". J. Phys. Chem. Lett. 11 (16): 6914–6920. doi:10.1021/acs.jpclett.0c01748. PMC 7450658. PMID 32787203.
- ^ Kwapiszewska, Karina; Kalwarczyk, Tomasz; Michalska, Bernadeta; Szczepański, Krzysztof; Szymański, Jędrzej; Patalas-Krawczyk, Paulina; Andryszewski, Tomasz; Iwan, Michalina; Duszyński, Jerzy; Hołyst, Robert (2019). "Determination of oligomerization state of Drp1 protein in living cells at nanomolar concentrations". Scientific Reports. 9 (1): 5906. Bibcode:2019NatSR...9.5906K. doi:10.1038/s41598-019-42418-0. PMC 6459820. PMID 30976093.
- ^ Kwapiszewska, Karina; Kalwarczyk, Tomasz; Michalska, Bernadeta; Szczepański, Krzysztof; Szymański, Jędrzej; Patalas-Krawczyk, Paulina; Andryszewski, Tomasz; Iwan, Michalina; Duszyński, Jerzy; Hołyst, Robert (2019). "Determination of oligomerization state of Drp1 protein in living cells at nanomolar concentrations". Scientific Reports. 9 (1): 5906. Bibcode:2019NatSR...9.5906K. doi:10.1038/s41598-019-42418-0. PMC 6459820. PMID 30976093.
- ^ Magde, D.; Elson, E. L.; Webb, W. W. (1972). "Thermodynamic fluctuations in a reacting system: Measurement by fluorescence correlation spectroscopy". Phys Rev Lett. 29 (11): 705–708. Bibcode:1972PhRvL..29..705M. doi:10.1103/physrevlett.29.705.
- ^ Ehrenberg, M.; Rigler, R. (1974). "Rotational brownian motion and fluorescence intensity fluctuations". Chem Phys. 4 (3): 390–401. Bibcode:1974CP......4..390E. doi:10.1016/0301-0104(74)85005-6.
- ^ Elson, E. L.; Magde, D. "Fluorescence correlation spectroscopy I. Conceptual basis and theory, (1974)". Biopolymers. 13: 1–27. doi:10.1002/bip.1974.360130102. S2CID 97201376.
- ^ a b Magde, D.; Elson, E. L.; Webb, W. W. (1974). "Fluorescence correlation spectroscopy II. An experimental realization". Biopolymers. 13 (1): 29–61. doi:10.1002/bip.1974.360130103. PMID 4818131. S2CID 2832069.
- ^ Qian, H.; Elson, E. L. (1991). "Analysis of confocal laser-microscope optics for 3-D fluorescence correlation spectroscopy". Applied Optics. 30 (10): 1185–1195. Bibcode:1991ApOpt..30.1185Q. doi:10.1364/AO.30.001185. PMID 20582127.
- ^ Palmer, A. G.; Thompson, N. L. (1989). "High-order fluorescence fluctuation analysis of model protein clusters". Proc Natl Acad Sci U S A. 86 (16): 6148–6152. Bibcode:1989PNAS...86.6148P. doi:10.1073/pnas.86.16.6148. PMC 297794. PMID 2548201.
- ^ Qian, H.; Elson, E. L. (1990). "Distribution of molecular aggregation by analysis of fluctuation moments". Proc Natl Acad Sci U S A. 87 (14): 5479–5483. Bibcode:1990PNAS...87.5479Q. doi:10.1073/pnas.87.14.5479. PMC 54348. PMID 2371284.
- ^ Thompson N L 1991 Topics in Fluorescence Spectroscopy Techniques vol 1, ed J R Lakowicz (New York: Plenum) pp 337–78
- ^ Rigler, R, Ü. Mets1, J. Widengren and P. Kask. "Fluorescence correlation spectroscopy with high count rate and low background: analysis of translational diffusion. European Biophysics Journal (1993) 22(3), 159.
- ^ Eigen, M.; Rigler, M. (1994). "Sorting single molecules: application to diagnostics and evolutionary biotechnology". Proc. Natl. Acad. Sci. USA. 91 (13): 5740–5747. Bibcode:1994PNAS...91.5740E. doi:10.1073/pnas.91.13.5740. PMC 44073. PMID 7517036.
- ^ Rigler, M (1995). "Fluorescence correlations, single molecule detection and large number screening. Applications in biotechnology". J. Biotechnol. 41 (2–3): 177–186. doi:10.1016/0168-1656(95)00054-t. PMID 7544589.
- ^ Krichevsky, O.; Bonnet, G. (2002). "Fluorescence correlation spectroscopy: the technique and its applications". Rep. Prog. Phys. 65 (2): 251–297. Bibcode:2002RPPh...65..251K. doi:10.1088/0034-4885/65/2/203. S2CID 49429529.
- ^ Medina, M. A.; Schwille, P. (2002). "Fluorescence correlation spectroscopy for the detection and study of single molecules in biology". BioEssays. 24 (8): 758–764. doi:10.1002/bies.10118. PMID 12210537. S2CID 3860264.
- ^ Mayboroda, O. A.; van Remoortere, A.; Tanke, H. J.; Hokke, C. H.; Deelder, A. M. (2003). "A new approach for fluorescence correlation spectroscopy (FCS) based immunoassays". J. Biotechnol. 107 (2): 185–192. doi:10.1016/j.jbiotec.2003.10.007. PMID 14711501.
- ^ Hess, S.T.; Webb, W.W. (2002). "Focal volume optics and experimental artifacts in confocal fluorescence correlation spectroscopy". Biophys. J. 83 (4): 2300–2317. Bibcode:2002BpJ....83.2300H. doi:10.1016/s0006-3495(02)73990-8. PMC 1302318. PMID 12324447.
- ^ Höfling, F.; Bamberg, K.-U. & Franosch, T. (2011). "Anomalous transport resolved in space and time by fluorescence correlation spectroscopy". Soft Matter. 7 (4): 1358–1363. arXiv:1003.3762. Bibcode:2011SMat....7.1358H. doi:10.1039/C0SM00718H. S2CID 18905838.
- ^ Banks, D. S.; Fradin, C. (2005). "Anomalous diffusion of proteins due to molecular crowding". Biophys. J. 89 (5): 2960–2971. Bibcode:2005BpJ....89.2960B. doi:10.1529/biophysj.104.051078. PMC 1366794. PMID 16113107.
- ^ Sengupta, P.; Garai, K.; Balaji, J.; Periasamy, N.; Maiti, S. (2003). "Measuring Size Distribution in Highly Heterogeneous Systems with Fluorescence Correlation Spectroscopy". Biophys. J. 84 (3): 1977–1984. Bibcode:2003BpJ....84.1977S. doi:10.1016/s0006-3495(03)75006-1. PMC 1302767. PMID 12609900.
- ^ Kohler, R.H.; Schwille, P.; Webb, W.W.; Hanson, M.R. (2000). "Active protein transport through plastid tubules: velocity quantified by fluorescence correlation spectroscopy". J Cell Sci. 113 (22): 3921–3930. doi:10.1242/jcs.113.22.3921. PMID 11058079.
- ^ Widengren, J.; Mets; Rigler, R. (1995). "Fluorescence correlation spectroscopy of triplet states in solution: a theoretical and experimental study". J. Chem. Phys. 99 (36): 13368–13379. doi:10.1021/j100036a009.
- ^ Berland, K. M. (2004). "Detection of specific DNA sequences using dual-color two-photon fluorescence correlation spectroscopy". J. Biotechnol. 108 (2): 127–136. doi:10.1016/j.jbiotec.2003.11.006. PMID 15129721.
- ^ a b c d Müller, C.B.; Loman, A.; Pacheco, V.; Koberling, F.; Willbold, D.; Richtering, W.; Enderlein, J. (2008). "Precise measurement of diffusion by multi-color dual-focus fluorescence correlation spectroscopy". EPL. 83 (4): 46001. Bibcode:2008EL.....8346001M. doi:10.1209/0295-5075/83/46001. S2CID 123509143.
- ^ Wang, F.; Shi, Y.; Luo, S.; Chen, Y.; Zhao, J. (2012). "Conformation transition of Poly(N-isopropylacrylamide) Single Chains in Its Cononsolvency Process: A Study by Fluorescence Correlation Spectroscopy and Scaling Analysis. (2012)". Macromolecules. 45 (22): 9196–9204. Bibcode:2012MaMol..45.9196W. doi:10.1021/ma301780f. S2CID 94553710.
- ^ a b Pristinski, D.; Kozlovskaya, V.; Sukhishvili, S. A. (2005). "Fluorescence correlation spectroscopy studies of diffusion of a weak polyelectrolyte in aqueous solutions". J. Chem. Phys. 122 (1): 014907. Bibcode:2005JChPh.122a4907P. doi:10.1063/1.1829255. PMID 15638700.
- ^ Widengren, J.; Schwille, P. (2000). "Characterization of photoinduced isomerization and back-isomerization of the cyanine dye Cy5 by fluorescence correlation spectroscopy. (2000)". J. Phys. Chem. A. 104 (27): 6416–6428. Bibcode:2000JPCA..104.6416W. doi:10.1021/jp000059s.
- ^ Loman, A.; Dertinger, T.; Koberling, F.; Enderlein, J. (2008). "Comparison of optical saturation effects in conventional and dual-focus fluorescence correlation spectroscopy (2008)". Chem. Phys. Lett. 459 (1): 18–21. Bibcode:2008CPL...459...18L. doi:10.1016/j.cplett.2008.05.018.
- ^ Petráaek; Schwille, P. (2008). "Precise Measurement of Diffusion Coefficients using Scanning Fluorescence Correlation Spectroscopy". Biophys. J. 94 (4): 1437–1448. Bibcode:2008BpJ....94.1437P. doi:10.1529/biophysj.107.108811. PMC 2212689. PMID 17933881.
- ^ Wawrezinieck et al. (2005) Biophys J.
- ^ Lenne et al. (2006) EMBO J.
- ^ Guia et al. (2011) Sci Signal.
- ^ Ruprecht et al. (2011) Biophys J.
- ^ Billaudeau et al. (2013) Methods In Enzymology
- ^ Masuda et al. (2005) Biophys J.
- ^ Humpolıckova et al. (2006) Biophys J.
- ^ Wenger et al. (2007) Biophys J.
- ^ Eggeling et al. (2009) Nature
- ^ Meseth, U.; Wohland, T.; Rigler, R.; Vogel, H. (1999). "Resolution of fluorescence correlation measurements. (1999)". Biophys. J. 76 (3): 1619–1631. Bibcode:1999BpJ....76.1619M. doi:10.1016/s0006-3495(99)77321-2. PMC 1300138. PMID 10049342.
- ^ Digman, M. A.; Dalal, R.; Horwitz, A. F.; Gratton, E. (2008). "Mapping the number of molecules and brightness in the laser scanning microscope". Biophys. J. 94 (6): 2320–2332. Bibcode:2008BpJ....94.2320D. doi:10.1529/biophysj.107.114645. PMC 2257897. PMID 18096627.
- ^ Chen, Y.; Müller, J. D.; So, P. T. C.; Gratton, E. (1999). "The photon counting histogram in fluorescence fluctuation spectroscopy". Biophys. J. 77 (1): 553–567. Bibcode:1999BpJ....77..553C. doi:10.1016/s0006-3495(99)76912-2. PMC 1300352. PMID 10388780.
- ^ Kask, P.; Palo, K.; Ullmann, D.; Gall, K. (1999). "Fluorescence-intensity distribution analysis and its application in biomolecular detection technology". Proc. Natl. Acad. Sci. U.S.A. 96 (24): 13756–13761. Bibcode:1999PNAS...9613756K. doi:10.1073/pnas.96.24.13756. PMC 24137. PMID 10570145.
- ^ Müller, J. D. (2004). "Cumulant analysis in fluorescence fluctuation spectroscopy". Biophys. J. 86 (6): 3981–3992. Bibcode:2004BpJ....86.3981M. doi:10.1529/biophysj.103.037887. PMC 1304299. PMID 15189894.
- ^ Godin, Antoine (April 26, 2011). "Revealing protein oligomerization and densities in situ using spatial intensity distribution analysis". PNAS. 108 (17): 7010–7015. Bibcode:2011PNAS..108.7010G. doi:10.1073/pnas.1018658108. PMC 3084122. PMID 21482753.
- ^ Içbilir, Ali (2021). "Determination of G-protein–coupled receptor oligomerization by molecular brightness analyses in single cells" (PDF). Nature Protocols. 16 (3): 1419–1451. doi:10.1038/s41596-020-00458-1. PMID 33514946. S2CID 231768809.
- ^ Qian, H.; Elson, E.L. (1990). "On the analysis of high order moments of fluorescence fluctuations". Biophys. J. 57 (2): 375–380. Bibcode:1990BpJ....57..375Q. doi:10.1016/s0006-3495(90)82539-x. PMC 1280678. PMID 2317556.
- ^ Remaut, K.; Lucas, B.; Braeckmans, K.; Sanders, N.N.; Smedt, S.C. De; Demeester, J. (2005). "FRET-FCS as a tool to evaluate the stability of oligonucleotide drugs after intracellular delivery". J Control Rel. 103 (1): 259–271. doi:10.1016/j.jconrel.2004.11.019. PMID 15710516.
- ^ Mashaghi, A.; et al. (2008). "Characterization of Protein Dynamics in Asymmetric Cell Division by Scanning Fluorescence Correlation Spectroscopy". Biophysical Journal. 95 (11): 5476–5486. Bibcode:2008BpJ....95.5476P. doi:10.1529/biophysj.108.135152. PMC 2586573. PMID 18805921.
- ^ Digman, M.A.; Sengupta, P.; Wiseman, P.W.; Brown, C.M.; Horwitz, A.R.; Gratton, E. (2005). "Fluctuation Correlation Spectroscopy with a Laser-Scanning Microscope: Exploiting the Hidden Time Structure". Biophys. J. 88 (5): L33–36. Bibcode:2005BpJ....88L..33D. doi:10.1529/biophysj.105.061788. PMC 1305524. PMID 15792971.
- ^ Skinner, J.P.; Chen, Y.; Mueller, J.D. (2005). "Position-Sensitive Scanning Fluorescence Correlation Spectroscopy". Biophys. J. 89 (2): 1288–1301. Bibcode:2005BpJ....89.1288S. doi:10.1529/biophysj.105.060749. PMC 1366613. PMID 15894645.
- ^ Ruan, Q.; Cheng, M.A.; Levi, M.; Gratton, E.; Mantulin, W.W. (2004). "Spatial-temporal studies of membrane dynamics: scanning fluorescence correlation spectroscopy (SFCS)". Biophys. J. 87 (2): 1260–1267. Bibcode:2004BpJ....87.1260R. doi:10.1529/biophysj.103.036483. PMC 1304464. PMID 15298928.
- ^ Berglund, A.; Mabuchi, H. (2005). "Tracking-FCS: Fluorescence correlation spectroscopy of individual particles" (PDF). Opt. Express. 13 (20): 8069–8082. Bibcode:2005OExpr..13.8069B. doi:10.1364/opex.13.008069. PMID 19498837.
- ^ Sisan, D.R.; Arevalo, R.; Graves, C.; McAllister, R.; Urbach, J.S. (2006). "Spatially resolved fluorescence correlation spectroscopy using a spinning disk confocal microscope". Biophysical Journal. 91 (11): 4241–4252. Bibcode:2006BpJ....91.4241S. doi:10.1529/biophysj.106.084251. PMC 1635679. PMID 16950838.
- ^ Kannan, B.; Guo, L.; Sudhaharan, T.; Ahmed, S.; Maruyama, I.; Wohland, T. (2007). "Spatially resolved total internal reflection fluorescence correlation microscopy using an electron multiplying charge-coupled device camera". Analytical Chemistry. 79 (12): 4463–4470. doi:10.1021/ac0624546. PMID 17489557.
- ^ Wachsmuth, M.; Waldeck, W.; Langowski, J. (2000). "Anomalous diffusion of fluorescent probes inside living cell nuclei investigated by spatially resolved fluorescence correlation spectroscopy". J. Mol. Biol. 298 (4): 677–689. doi:10.1006/jmbi.2000.3692. PMID 10788329. S2CID 21791229.
- ^ a b c Hebert, B.; Constantino, S.; Wiseman, P. W. (2005). "Spatio-temporal image correlation spectroscopy (STICS): theory, verification and application to protein velocity mapping in living CHO cells". Biophys. J. 88 (5): 3601–3614. Bibcode:2005BpJ....88.3601H. doi:10.1529/biophysj.104.054874. PMC 1305507. PMID 15722439.
- ^ a b Wiseman, P. W.; Squier, J. A.; Ellisman, M. H.; Wilson, K. R. (2000). "Two-photon video rate image correlation spectroscopy (ICS) and image cross-correlation spectroscopy (ICCS)". J. Microsc. 200 (Pt 1): 14–25. doi:10.1046/j.1365-2818.2000.00736.x. PMID 11012824. S2CID 6554931.
- ^ Petersen, N. O.; Wiseman, P. W.; Seger, O.; Magnusson, K. E. (1993). "Quantitation of membrane receptor distributions by image correlation spectroscopy: concept and application". Biophys. J. 65 (3): 1135–1146. Bibcode:1993BpJ....65.1135P. doi:10.1016/S0006-3495(93)81173-1. PMC 1225831. PMID 8241393.
- ^ Kolin, D.L.; Ronis, D.; Wiseman, P.W. (2006). "k-Space Image Correlation Spectroscopy: A Method for Accurate Transport Measurements Independent of Fluorophore Photophysics". Biophys. J. 91 (8): 3061–3075. Bibcode:2006BpJ....91.3061K. doi:10.1529/biophysj.106.082768. PMC 1578478. PMID 16861272.
- ^ Semrau, S.; Schmidt, T. (2007). "Particle Image Correlation Spectroscopy (PICS): Retrieving Nanometer-Scale Correlations from High-Density Single-Molecule Position Data". Biophys. J. 92 (2): 613–621. Bibcode:2007BpJ....92..613S. doi:10.1529/biophysj.106.092577. PMC 1751376. PMID 17085496.
- ^ Semrau, S.; Holtzer, L.; Gonzalez-Gaitan, M.; Schmidt, T. (2011). "Quantification of Biological Interactions with Particle Image Cross-Correlation Spectroscopy (PICCS)". Biophys. J. 100 (7): 1810–1818. Bibcode:2011BpJ...100.1810S. doi:10.1016/j.bpj.2010.12.3746. PMC 3072609. PMID 21463595.
- ^ a b c Kisley, L.; Higgins, D.; Weiss, S.; Landes, C.F. (2015). "Characterization of Porous Materials by Fluorescence Correlation Spectroscopy Super-resolution Optical Fluctuation Imaging". ACS Nano. 9 (9): 9158–9166. doi:10.1021/acsnano.5b03430. PMC 10706734. PMID 26235127.
- ^ Dutta, C.; Bishop, L. D. C.; Landes, C.F. (2020). "Imaging Switchable Protein Interactions with an Active Porous Polymer Support". J. Phys. Chem. B. 124 (22): 4412–4420. doi:10.1021/acs.jpcb.0c01807. PMID 32441098. S2CID 218836568.
- ^ Shayegan, M.; Michnick, S. W.; Leslie, S. L. (2019). "Probing Inhomogeneous Diffusion in the Microenvironments of Phase-Separated Polymers under Confinement". J. Am. Chem. Soc. 141 (19): 7751–7757. doi:10.1021/jacs.8b13349. PMID 31017394. S2CID 129941554.
- ^ Lieto, A.M.; Thompson, N.L. (2004). "Total Internal Reflection with Fluorescence Correlation Spectroscopy: Nonfluorescent Competitors". Biophys. J. 87 (2): 1268–1278. Bibcode:2004BpJ....87.1268L. doi:10.1529/biophysj.103.035030. PMC 1304465. PMID 15298929.
- ^ Capoulade, J.; Wachsmuth, M.; Hufnagel, L.; Knop, M. (September 2011). "Quantitative fluorescence imaging of protein diffusion and interaction in living cells". Nature Biotechnology. 29 (9): 835–839. doi:10.1038/nbt.1928. PMID 21822256. S2CID 10493584.
- ^ Sprague, B.L.; McNally, J.G. (2005). "FRAP analysis of binding: proper and fitting". Trends in Cell Biology. 15 (2): 84–91. doi:10.1016/j.tcb.2004.12.001. PMID 15695095.
- ^ Barulin, Aleksandr; Claude, Jean-Benoît; Patra, Satyajit; Bonod, Nicolas; Wenger, Jérôme (9 October 2019). "Deep Ultraviolet Plasmonic Enhancement of Single Protein Autofluorescence in Zero-Mode Waveguides". Nano Letters. 19 (10): 7434–7442. arXiv:1909.08227. Bibcode:2019NanoL..19.7434B. doi:10.1021/acs.nanolett.9b03137. PMID 31526002. S2CID 202660648.
- ^ Barulin, Aleksandr; Roy, Prithu; Claude, Jean-Benoît; Wenger, Jérôme (5 April 2022). "Ultraviolet optical horn antennas for label-free detection of single proteins". Nature Communications. 13 (1): 1842. arXiv:2204.02807. Bibcode:2022NatCo..13.1842B. doi:10.1038/s41467-022-29546-4. PMC 8983662. PMID 35383189.
- ^ Roy, Prithu; Claude, Jean-Benoît; Tiwari, Sunny; Barulin, Aleksandr; Wenger, Jérôme (5 January 2023). "Ultraviolet Nanophotonics Enables Autofluorescence Correlation Spectroscopy on Label-Free Proteins with a Single Tryptophan". Nano Letters. 23 (2): 497–504. arXiv:2301.01516. Bibcode:2023NanoL..23..497R. doi:10.1021/acs.nanolett.2c03797. PMID 36603115. S2CID 255416119.
- ^ Diaspro, A.; Robello, M. (1999). "Multi-photon Excitation Microscopy to Study Biosystems". European Microscopy and Analysis. 5: 5–7.
- ^ Bagatolli, L.A.; Gratton, E. (2000). "Two-photon fluorescence microscopy of coexisting lipid domains in giant unilamellar vesicles of binary phospholipid mixtures". Biophys J. 78 (1): 290–305. Bibcode:2000BpJ....78..290B. doi:10.1016/s0006-3495(00)76592-1. PMC 1300637. PMID 10620293.
- ^ Schwille, P.; Haupts, U.; Maiti, S.; Webb, W. (1999). "Molecular dynamics in living cells observed by fluorescence correlation spectroscopy with one- and two- photon excitation". Biophysical Journal. 77 (10): 2251–2265. Bibcode:1999BpJ....77.2251S. doi:10.1016/S0006-3495(99)77065-7. PMC 1300505. PMID 10512844.
- ^ Near Infrared Microspectroscopy, Fluorescence Microspectroscopy, Infrared Chemical Imaging and High Resolution Nuclear Magnetic Resonance Analysis of Soybean Seeds, Somatic Embryos and Single Cells., Baianu, I.C. et al. 2004., In Oil Extraction and Analysis., D. Luthria, Editor pp.241–273, AOCS Press., Champaign, IL.
- ^ Single Cancer Cell Detection by Near Infrared Microspectroscopy, Infrared Chemical Imaging and Fluorescence Microspectroscopy.2004.I. C. Baianu, D. Costescu, N. E. Hofmann and S. S. Korban, q-bio/0407006 (July 2004)
Further reading edit
- Rigler R. and Widengren J. (1990). Ultrasensitive detection of single molecules by fluorescence correlation spectroscopy, BioScience (Ed. Klinge & Owman) p. 180
- Oehlenschläger, F.; Schwille, P.; Eigen, M. (1996). "Detection of HIV-1 RNA by nucleic acid sequence-based amplification combined with fluorescence correlation spectroscopy". Proc. Natl. Acad. Sci. USA. 93 (23): 12811–12816. Bibcode:1996PNAS...9312811O. doi:10.1073/pnas.93.23.12811. PMC 24002. PMID 8917501.
External links edit
- Haustein, Elke; Schwille, Petra (2004). "Single-molecule spectroscopic methods". Current Opinion in Structural Biology. 14 (5): 531–540. doi:10.1016/j.sbi.2004.09.004. hdl:11858/00-001M-0000-0029-D76C-C. PMID 15465312.
- FCS Classroom
- Stowers Institute FCS Tutorial
- Cell Migration Consortium FCS Tutorial Archived 2010-09-24 at the Wayback Machine
- Fluorescence Correlation Spectroscopy (FCS) (Becker & Hickl GmbH, web page)