


Summary
In polymer chemistry, living polymerization is a form of chain growth polymerization where the ability of a growing polymer chain to terminate has been removed.[1][2] This can be accomplished in a variety of ways. Chain termination and chain transfer reactions are absent and the rate of chain initiation is also much larger than the rate of chain propagation. The result is that the polymer chains grow at a more constant rate than seen in traditional chain polymerization and their lengths remain very similar (i.e. they have a very low polydispersity index). Living polymerization is a popular method for synthesizing block copolymers since the polymer can be synthesized in stages, each stage containing a different monomer. Additional advantages are predetermined molar mass and control over end-groups.

Living polymerization is desirable because it offers precision and control in macromolecular synthesis. This is important since many of the novel/useful properties of polymers result from their microstructure and molecular weight. Since molecular weight and dispersity are less controlled in non-living polymerizations, this method is more desirable for materials design[3][4]
In many cases, living polymerization reactions are confused or thought to be synonymous with controlled polymerizations. While these polymerization reactions are very similar, there is a distinction between the definitions of these two reactions. While living polymerizations are defined as polymerization reactions where termination or chain transfer is eliminated, controlled polymerization reactions are reactions where termination is suppressed, but not eliminated, through the introduction of a dormant state of the polymer.[3][4] However, this distinction is still up for debate in the literature.
The main living polymerization techniques are:
- Living anionic polymerization
- Living cationic polymerization
- Living ring-opening metathesis polymerization
- Living free radical polymerization
- Living chain-growth polycondensations
History edit
Living polymerization was demonstrated by Michael Szwarc in 1956 in the anionic polymerization of styrene with an alkali metal / naphthalene system in tetrahydrofuran (THF). Szwarc showed that electron transfer occurred from radical anion of naphthalene to styrene. The initial radical anion of styrene converts to a dianion (or equivalently disodio-) species, which rapidly added styrene to form a "two – ended living polymer." An important aspect of his work, Szwarc employed the aprotic solvent tetrahydrofuran, which dissolves but is otherwise unreactive toward the organometallic intermediates. After initial addition of monomer to the initiator system, the viscosity increased (due to increased polymer chain growth), but eventually cease after depletion of monomer concentration. However, he found that addition of more monomer caused an increase in viscosity, indicating growth of the polymer chain, and thus concluded that the polymer chains had never been terminated.[5] This was a major step in polymer chemistry, since control over when the polymer was quenched, or terminated, was generally not a controlled step. With this discovery, the list of potential applications expanded dramatically.[6]
Today, living polymerizations are used widely in the production of many types of polymers or plastics. For instance, poly(phthalaldehyde) polymer, first developed in 1967, can be synthesized via both living cationic and living anionic polymerization reactions producing both the cyclic or linear form of the polymer respectively. The approach offers control of the chemical makeup of the polymer and, thus, the structural and electronic properties of the material. This level of control rarely exists in non-living polymerization reactions.[4][7]
Fast rate of initiation: low polydispersity edit


One of the key characteristics of a living polymerization is that the chain termination and transfer reactions are essentially eliminated from the four elementary reactions of chain-growth polymerization leaving only initiation and (chain) propagation reactions.
A key characteristic of living polymerization is that the rate of initiation (meaning the dormant chemical species generates the active chain propagating species) is much faster than the rate of chain propagation. Thus all of the chains grow at the same rate (the rate of propagation).
The high rate of initiation (together with absence of termination) results in low (or narrow) polydispersity index (PDI), an indication of the broadness in the distribution of polymer chains.[8] The extended lifetime of the propagating chain allowing for co-block polymer formation and end group functionalization to be performed on the living chain. These factors also allow predictable molecular weights, expressed as the number average molecular weight (Mn). For an ideal living system, assuming efficiency for generating active species is 100%, where each initiator generates only one active species the Kinetic chain length (average number of monomers the active species reacts with during its lifetime) at a given time can be estimated by knowing the concentration of monomer remaining. The number average molecular weight, Mn, increases linearly with percent conversion during a living polymerization
![{\displaystyle \ v={\frac {[M]_{0}-[M]}{[I]_{0}}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/db304320640f163330c3130db03ff9691e1a8ffa)
Techniques edit
Living anionic polymerization edit
As early as 1936, Karl Ziegler proposed that anionic polymerization of styrene and butadiene by consecutive addition of monomer to an alkyl lithium initiator occurred without chain transfer or termination. Twenty years later, living polymerization was demonstrated by Szwarc through the anionic polymerization of styrene in THF using sodium naphthalene as an initiator.[9][5][10]
The naphthalene anion initiates polymerization by reducing styrene to its radical anion, which dimerizes to the dilithiodiphenylbutane, which then initiates the polymerization. These experiments relied on Szwarc's ability to control the levels of impurities which would destroy the highly reactive organometallic intermediates.
Living α-olefin polymerization edit
α-olefins can be polymerized through an anionic coordination polymerization in which the metal center of the catalyst is considered the counter cation for the anionic end of the alkyl chain (through a M-R coordination). Ziegler-Natta initiators were developed in the mid-1950s and are heterogeneous initiators used in the polymerization of alpha-olefins. Not only were these initiators the first to achieve relatively high molecular weight poly(1-alkenes) (currently the most widely produced thermoplastic in the world PE(Polyethylene) and PP (Polypropylene)[11] but the initiators were also capable of stereoselective polymerizations which is attributed to the chiral Crystal structure of the heterogeneous initiator.[4] Due to the importance of this discovery Ziegler and Natta were presented with the 1963 Nobel Prize in chemistry. Although the active species formed from the Ziegler-Natta initiator generally have long lifetimes (on the scale of hours or longer) the lifetimes of the propagating chains are shortened due to several chain transfer pathways (Beta-Hydride elimination and transfer to the co-initiator) and as a result are not considered living.[4]
Metallocene initiators are considered as a type of Ziegler-Natta initiators due to the use of the two-component system consisting of a transition metal and a group I-III metal co-initiator (for example methylalumoxane (MAO) or other alkyl aluminum compounds). The metallocene initiators form homogeneous single site catalysts that were initially developed to study the impact that the catalyst structure had on the resulting polymers structure/properties; which was difficult for multi-site heterogeneous Ziegler-Natta initiators.[11] Owing to the discrete single site on the metallocene catalyst researchers were able to tune and relate how the ancillary ligand (those not directly involved in the chemical transformations) structure and the symmetry about the chiral metal center affect the microstructure of the polymer.[12] However, due to chain breaking reactions (mainly Beta-Hydride elimination) very few metallocene based polymerizations are known.[4]
By tuning the steric bulk and electronic properties of the ancillary ligands and their substituents a class of initiators known as chelate initiators (or post-metallocene initiators) have been successfully used for stereospecific living polymerizations of alpha-olefins. The chelate initiators have a high potential for living polymerizations because the ancillary ligands can be designed to discourage or inhibit chain termination pathways. Chelate initiators can be further broken down based on the ancillary ligands; ansa-cyclopentyadienyl-amido initiators, alpha-diimine chelates and phenoxy-imine chelates.[4]
- Ansa-cyclopentadienyl-amido (CpA) initiators

CpA initiators have one cyclopentadienyl substituent and one or more nitrogen substituents coordinated to the metal center (generally a Zr or Ti) (Odian). The dimethyl(pentamethylcyclopentyl)zirconium acetamidinate in figure___ has been used for a stereospecific living polymerization of 1-hexene at −10 °C. The resulting poly(1-hexene) was isotactic (stereochemistry is the same between adjacent repeat units) confirmed by 13C-NMR. The multiple trials demonstrated a controllable and predictable (from catalyst to monomer ratio) Mn with low Đ. The polymerization was further confirmed to be living by sequentially adding 2 portions of the monomer, the second portion was added after the first portion was already polymerized, and monitoring the Đ and Mn of the chain. The resulting polymer chains complied with the predicted Mn (with the total monomer concentration = portion 1 +2) and showed low Đ[13] suggesting the chains were still active, or living, as the second portion of monomer was added (5).
- α-diimine chelate initiators
α-diimine chelate initiators are characterized by having a diimine chelating ancillary ligand structure and which is generally coordinated to a late transition (i.e. Ni and Pd) metal center.

Brookhart et al. did extensive work with this class of catalysts and reported living polymerization for α-olefins[14] and demonstrated living α-olefin carbon monoxide alternating copolymers.[15]
Living cationic polymerization edit
Monomers for living cationic polymerization are electron-rich alkenes such as vinyl ethers, isobutylene, styrene, and N-vinylcarbazole. The initiators are binary systems consisting of an electrophile and a Lewis acid. The method was developed around 1980 with contributions from Higashimura, Sawamoto and Kennedy. Typically, generating a stable carbocation for a prolonged period of time is difficult, due to the possibility for the cation to be quenched by a β-protons attached to another monomer in the backbone, or in a free monomer. Therefore, a different approach is taken[3][4][16]

In this example, the carbocation is generated by the addition of a Lewis acid (co-initiator, along with the halogen "X" already on the polymer – see figure), which ultimately generates the carbocation in a weak equilibrium. This equilibrium heavily favors the dormant state, thus leaving little time for permanent quenching or termination by other pathways. In addition, a weak nucleophile (Nu:) can also be added to reduce the concentration of active species even further, thus keeping the polymer "living".[3][4][16] However, it is important to note that by definition, the polymers described in this example are not technically living due to the introduction of a dormant state, as termination has only been decreased, not eliminated (though this topic is still up for debate). But, they do operate similarly, and are used in similar applications to those of true living polymerizations.
Living ring-opening metathesis polymerization edit
Given the right reaction conditions ring-opening metathesis polymerization (ROMP) can be rendered living. The first such systems were described by Robert H. Grubbs in 1986 based on norbornene and Tebbe's reagent and in 1978 Grubbs together with Richard R. Schrock describing living polymerization with a tungsten carbene complex.[17]
Generally, ROMP reactions involve the conversion of a cyclic olefin with significant ring-strain (>5 kcal/mol), such as cyclobutene, norbornene, cyclopentene, etc., to a polymer that also contains double bonds. The important thing to note about ring-opening metathesis polymerizations is that the double bond is usually maintained in the backbone, which can allow it to be considered "living" under the right conditions.[18]
For a ROMP reaction to be considered "living", several guidelines must be met:[18]
- Fast and complete initiation of the monomer. This means that the rate at which an initiating agent activates the monomer for polymerization, must happen very quickly.
- How many monomers make up each polymer (the degree of polymerization) must be related linearly to the amount of monomer you started with.
- The dispersity of the polymer must be < 1.5. In other words, the distribution of how long your polymer chains are in your reaction must be very low.
With these guidelines in mind, it allows you to create a polymer that is well controlled both in content (what monomer you use) and properties of the polymer (which can be largely attributed to polymer chain length). It is important to note that living ring-opening polymerizations can be anionic or cationic.

Because living polymers have had their termination ability removed, this means that once your monomer has been consumed, the addition of more monomer will result in the polymer chains continuing to grow until all of the additional monomer is consumed. This will continue until the metal catalyst at the end of the chain is intentionally removed by the addition of a quenching agent. As a result, it may potentially allow one to create a block or gradient copolymer fairly easily and accurately. This can lead to a high ability to tune the properties of the polymer to a desired application (electrical/ionic conduction, etc.)[4][18]
"Living" free radical polymerization edit
Starting in the 1970s several new methods were discovered which allowed the development of living polymerization using free radical chemistry. These techniques involved catalytic chain transfer polymerization, iniferter mediated polymerization, stable free radical mediated polymerization (SFRP), atom transfer radical polymerization (ATRP), reversible addition-fragmentation chain transfer (RAFT) polymerization, and iodine-transfer polymerization.
In "living" radical polymerization (or controlled radical polymerization (CRP)) the chain breaking pathways are severely depressed when compared to conventional radical polymerization (RP) and CRP can display characteristics of a living polymerization. However, since chain termination is not absent, but only minimized, CRP technically does not meet the requirements imposed by IUPAC for a living polymerization (see introduction for IUPAC definition). This issue has been up for debate the view points of different researchers can be found in a special issue of the Journal of Polymer Science titled Living or Controlled ?. The issue has not yet been resolved in the literature so it is often denoted as a "living" polymerization, quasi-living polymerization, pseudo-living and other terms to denote this issue.
There are two general strategies employed in CRP to suppress chain breaking reactions and promote fast initiation relative to propagation. Both strategies are based on developing a dynamic equilibrium amongst an active propagating radical and a dormant species.[19]
The first strategy involves a reversible trapping mechanism in which the propagating radical undergoes an activation/deactivation (i.e. Atom-transfer radical-polymerization) process with a species X. The species X is a persistent radical, or a species that can generate a stable radical, that cannot terminate with itself or propagate but can only reversibly "terminate" with the propagating radical (from the propagating polymer chain) P*. P* is a radical species that can propagate (kp) and irreversibly terminate (kt) with another P*. X is normally a nitroxide (i.e. TEMPO used in Nitroxide Mediated Radical Polymerization) or an organometallic species. The dormant species (Pn-X) can be activated to regenerate the active propagating species (P*) spontaneously, thermally, using a catalyst and optically.[19][20]
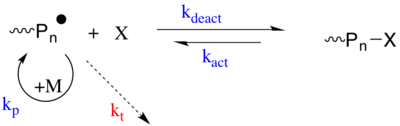
The second strategy is based on a degenerative transfer (DT) of the propagating radical between transfer agent that acts as a dormant species (i.e. Reversible addition−fragmentation chain-transfer polymerization). The DT based CRP's follow the conventional kinetics of radical polymerization, that is slow initiation and fast termination, but the transfer agent (Pm-X or Pn-X) is present in a much higher concentration compared to the radical initiator. The propagating radical species undergoes a thermally neutral exchange with the dormant transfer agent through atom transfer, group transfer or addition fragment chemistry.[19]
Living chain-growth polycondensations edit
Chain growth polycondensation polymerizations were initially developed under the premise that a change in substituent effects of the polymer, relative to the monomer, causes the polymers end group to be more reactive this has been referred to as "reactive intermediate polycondensation". The essential result is monomers preferentially react with the activated polymer end groups over reactions with other monomers. This preferred reactivity is the fundamental difference when categorizing a polymerization mechanism as chain-growth as opposed to step-growth in which the monomer and polymer chain end group have equal reactivity (the reactivity is uncontrolled). Several strategies were employed to minimize monomer-monomer reactions (or self-condensation) and polymerizations with low D and controllable Mn have been attained by this mechanism for small molecular weight polymers.[21] However, for high molecular weight polymer chains (i.e. small initiator to monomer ratio) the Mn is not easily to controlled, for some monomers, since self-condensation between monomers occurred more frequently due to the low propagating species concentration.[21]
Catalyst-transfer polycondensation edit
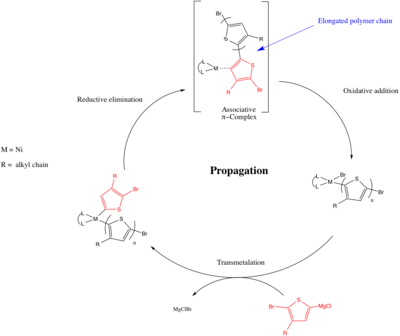
Catalyst transfer polycondensation (CTP) is a chain-growth polycondensation mechanism in which the monomers do not directly react with one another and instead the monomer will only react with the polymer end group through a catalyst-mediated mechanism.[21] The general process consists of the catalyst activating the polymer end group followed by a reaction of the end group with a 2nd incoming monomer. The catalyst is then transferred to the elongated chain while activating the end group (as shown below).[22]

Catalyst transfer polycondensation allows for the living polymerization of π-conjugated polymers and was discovered by Tsutomu Yokozawa in 2004[22] and Richard McCullough.[23] In CTP the propagation step is based on organic cross coupling reactions (i.e. Kumada coupling, Sonogashira coupling, Negishi coupling) top form carbon carbon bonds between difunctional monomers. When Yokozawa and McCullough independently discovered the polymerization using a metal catalyst to couple a Grignard reagent with an organohalide making a new carbon-carbon bond. The mechanism below shows the formation of poly(3-alkylthiophene) using a Ni initiator (Ln can be 1,3-Bis(diphenylphosphino)propane (dppp)) and is similar to the conventional mechanism for Kumada coupling involving an oxidative addition, a transmetalation and a reductive elimination step. However, there is a key difference, following reductive elimination in CTP, an associative complex is formed (which has been supported by intra-/intermolecular oxidative addition competition experiments[7]) and the subsequent oxidative addition occurs between the metal center and the associated chain (an intramolecular pathway). Whereas in a coupling reaction the newly formed alkyl/aryl compound diffuses away and the subsequent oxidative addition occurs between an incoming Ar–Br bond and the metal center. The associative complex is essential to for polymerization to occur in a living fashion since it allows the metal to undergo a preferred intramolecular oxidative addition and remain with a single propagating chain (consistent with chain-growth mechanism), as opposed to an intermolecular oxidative addition with other monomers present in the solution (consistent with a step-growth, non-living, mechanism).[24][25] The monomer scope of CTP has been increasing since its discovery and has included poly(phenylene)s, poly(fluorine)s, poly(selenophene)s and poly(pyrrole)s.[24][25]
Living group-transfer polymerization edit
Group-transfer polymerization also has characteristics of living polymerization.[26] It is applied to alkylated methacrylate monomers and the initiator is a silyl ketene acetal. New monomer adds to the initiator and to the active growing chain in a Michael reaction. With each addition of a monomer group the trimethylsilyl group is transferred to the end of the chain. The active chain-end is not ionic as in anionic or cationic polymerization but is covalent. The reaction can be catalysed by bifluorides and bioxyanions such as tris(dialkylamino)sulfonium bifluoride or tetrabutyl ammonium bibenzoate. The method was discovered in 1983 by Owen Webster[27] and the name first suggested by Barry Trost.
Applications edit
Living polymerizations are used in the commercial synthesis of many polymers.
Copolymer synthesis and applications edit
Copolymers are polymers consisting of multiple different monomer species, and can be arranged in various orders, three of which are seen in the figure below.

While there exist others (alternating copolymers, graft copolymers, and stereoblock copolymers), these three are more common in the scientific literature.[3] In addition, block copolymers can exist as many types, including triblock (A-B-A), alternating block (A-B-A-B-A-B), etc.
Of these three types, block and gradient copolymers are commonly synthesized through living polymerizations, due to the ease of control living polymerization provides. Copolymers are highly desired due to the increased flexibility of properties a polymer can have compared to their homopolymer counterparts. The synthetic techniques used range from ROMP to generic anionic or cationic living polymerizations.[3][4]
Copolymers, due to their unique tunability of properties, can have a wide range of applications. One example (of many) is nano-scale lithography using block copolymers. One used frequently is a block copolymer made of polystyrene and poly(methyl methacrylate) (abbreviated PS-b-PMMA). This copolymer, upon proper thermal and processing conditions, can form cylinders on the order of a few tens of nanometers in diameter of PMMA, surrounded by a PS matrix. These cylinders can then be etched away under high exposure to UV light and acetic acid, leaving a porous PS matrix.[28][29][30]
The unique property of this material is that the size of the pores (or the size of the PMMA cylinders) can be easily tuned by the ratio of PS to PMMA in the synthesis of the copolymer. This can be easily tuned due to the easy control given by living polymerization reactions, thus making this technique highly desired for various nanoscale patterning of different materials for applications to catalysis, electronics, etc.
References edit
- ^ Halasa, A. F. (1981). "Recent Advances in Anionic Polymerization". Rubber Chemistry and Technology. 54 (3): 627–640. doi:10.5254/1.3535823.
- ^ Moad, Graeme and Solomon, David H. (2006) The Chemistry of Radical Polymerization. 2nd ed. Elsevier. ISBN 0-08-044286-2
- ^ a b c d e f Cowie, J.M.G. (2007). Polymers chemistry and physics of modern materials (3rd ed / J.M.G. Cowie and Valeria Arrighi ed.). Boca Raton: Taylor & Francis. ISBN 9780849398131.
- ^ a b c d e f g h i j k l Odian, George (2004). Principles of polymerization (4. ed.). Hoboken, NJ: Wiley-Interscience. ISBN 978-0471274001.
- ^ a b Szwarc, M.; Levy, M.; Milkovich, R. (1956). "Polymerization Initiated by Electron Transfer to Monomer. A New Method of Formation of Block Polymers1". Journal of the American Chemical Society. 78 (11): 2656–2657. doi:10.1021/ja01592a101.
- ^ M. Szwarc (1956). ""Living" polymers". Nature. 178 (4543): 1168. doi:10.1038/1781168a0.
- ^ a b McNeil, Anne; Bryan, Zachary (2013). "Evidence for a preferential intramolecular oxidative addition in Ni-catalyzed cross-coupling reactions and their impact on chain-growth polymerizations". Chem. Sci. 4 (4): 1620–1624. doi:10.1039/C3SC00090G.
{{cite journal}}: CS1 maint: multiple names: authors list (link) - ^ "Living polymer". Gold Book. International Union of Pure and Applied Chemistry . doi:10.1351/goldbook.LT07156. Retrieved 4 January 2023.
- ^ Szwarc, M. (1956). "'Living' Polymers". Nature. 178 (4543): 1168–1169. Bibcode:1956Natur.178.1168S. doi:10.1038/1781168a0.
- ^ Tatemoto, Masayoshi and Nakagawa, Tsuneo "Segmented polymers containing fluorine and iodine and their production" U.S. patent 4,158,678. Priority date 30 June 1976.
- ^ a b Craver, C.; Carraher, C. (2000). Applied Polymer Science: 21st Century. Elsevier. pp. 1022–1023.
- ^ Coates, Geoffrey W. (April 2000). "Precise Control of Polyolefin Stereochemistry Using Single-Site Metal Catalysts". Chemical Reviews. 100 (4): 1223–1252. doi:10.1021/cr990286u. PMID 11749265.
- ^ Jayaratne, K.; Sita, L (2000). "Stereospecific Living Ziegler−Natta Polymerization of 1-Hexene". J. Am. Chem. Soc. 122 (5): 958–959. doi:10.1021/ja993808w.
- ^ Killian, C. M.; Tempel, D. J.; Johnson, L. K.; Brookhart, M. (1996). "Living Polymerization of α-Olefins Using NiII−α-Diimine Catalysts. Synthesis of New Block Polymers Based on α-Olefins". Journal of the American Chemical Society. 118 (46): 11664–11665. doi:10.1021/ja962516h.
- ^ Brookhart, M.; Rix, F. C.; Desimone, J. M.; Barborak, J. C. (1992). "Palladium(II) catalysts for living alternating copolymerization of olefins and carbon monoxide". Journal of the American Chemical Society. 114 (14): 5894–5895. doi:10.1021/ja00040a082.
- ^ a b Goethals, E; Duprez, F (2007). "Carbocationic polymerizations". Progress in Polymer Science. 32 (2): 220–246. doi:10.1016/j.progpolymsci.2007.01.001.
- ^ Schrock, R. R.; Feldman, J.; Cannizzo, L. F.; Grubbs, R. H. (1987). "Ring-opening polymerization of norbornene by a living tungsten alkylidene complex". Macromolecules. 20 (5): 1169–1172. Bibcode:1987MaMol..20.1169S. doi:10.1021/ma00171a053.
- ^ a b c Bielawski, Christopher W.; Grubbs, Robert H. (2007). "Living ring-opening metathesis polymerization". Progress in Polymer Science. 32 (1): 1–29. doi:10.1016/j.progpolymsci.2006.08.006.
- ^ a b c Braunecker, Wade A.; Matyjaszewski, Krzysztof (2007). "Controlled/living radical polymerization: Features, developments, and perspectives". Progress in Polymer Science. 32 (1): 93–146. doi:10.1016/j.progpolymsci.2006.11.002.
- ^ Matyjaszewski. "Features of Controlled "Living" Polymerization". Archived from the original on 14 March 2014.
- ^ a b c Yokozawa, T.; Yokoyama, A. (2007). "Chain-growth polycondensation: The living polymerization process in polycondensation". Progress in Polymer Science. 32: 147–172. doi:10.1016/j.progpolymsci.2006.08.001.
- ^ a b Miyakoshi, Ryo; Yokoyama, Akihiro; Yokozawa, Tsutomu (2005). "Catalyst-Transfer Polycondensation. Mechanism of Ni-Catalyzed Chain-Growth Polymerization Leading to Well-Defined Poly(3-hexylthiophene)". Journal of the American Chemical Society. 127 (49): 17542–17547. doi:10.1021/ja0556880. PMID 16332106.
- ^ Iovu, Mihaela Corina; Sheina, Elena E.; Gil, Roberto R.; McCullough, Richard D. (October 2005). "Experimental Evidence for the Quasi-"Living" Nature of the Grignard Metathesis Method for the Synthesis of Regioregular Poly(3-alkylthiophenes)". Macromolecules. 38 (21): 8649–8656. Bibcode:2005MaMol..38.8649I. CiteSeerX 10.1.1.206.3875. doi:10.1021/ma051122k.
- ^ a b Kiriy, Anton; Senkovskyy, Volodymyr; Sommer, Michael (4 October 2011). "Kumada Catalyst-Transfer Polycondensation: Mechanism, Opportunities, and Challenges". Macromolecular Rapid Communications. 32 (19): 1503–1517. doi:10.1002/marc.201100316. PMID 21800394.
- ^ a b Bryan, Zachary J.; McNeil, Anne J. (12 November 2013). "Conjugated Polymer Synthesis via Catalyst-Transfer Polycondensation (CTP): Mechanism, Scope, and Applications". Macromolecules. 46 (21): 8395–8405. Bibcode:2013MaMol..46.8395B. doi:10.1021/ma401314x. S2CID 101567648.
- ^ Davis, Fred J. (2004) Polymer chemistry: a practical approach. Oxford University Press. ISBN 978-0-19-850309-5 .
- ^ Webster, O. W.; Hertler, W. R.; Sogah, D. Y.; Farnham, W. B.; RajanBabu, T. V. (1983). "Group-transfer polymerization. 1. A new concept for addition polymerization with organosilicon initiators". J. Am. Chem. Soc. 105 (17): 5706–5708. doi:10.1021/ja00355a039.
- ^ In, Insik; La, Young-Hye; Park, Sang-Min; Nealey, Paul F.; Gopalan, Padma (August 2006). "Side-Chain-Grafted Random Copolymer Brushes as Neutral Surfaces for Controlling the Orientation of Block Copolymer Microdomains in Thin Films". Langmuir. 22 (18): 7855–7860. doi:10.1021/la060748g. PMID 16922574.
- ^ Liu, Yuanjun; Gong, Yanchun; He, Longbin; Xie, Bo; Chen, Xi; Han, Min; Wang, Guanghou (2010). "Formation of periodic nanoring arrays on self-assembled PS-b-PMMA film under rapid solvent-annealing". Nanoscale. 2 (10): 2065–8. Bibcode:2010Nanos...2.2065L. doi:10.1039/c0nr00207k. PMID 20820641.
- ^ Edwards, E. W.; Montague, M. F.; Solak, H. H.; Hawker, C. J.; Nealey, P. F. (4 August 2004). "Precise Control over Molecular Dimensions of Block-Copolymer Domains Using the Interfacial Energy of Chemically Nanopatterned Substrates". Advanced Materials. 16 (15): 1315–1319. doi:10.1002/adma.200400763. S2CID 95470388.
External links edit
- IUPAC Gold Book Definition
- Living Ziegler-Natta Polymerization Article
- Living polymers 50 years of evolution Article