


Summary
Molecular graphics is the discipline and philosophy of studying molecules and their properties through graphical representation.[1] IUPAC limits the definition to representations on a "graphical display device".[2] Ever since Dalton's atoms and Kekulé's benzene, there has been a rich history of hand-drawn atoms and molecules, and these representations have had an important influence on modern molecular graphics.
Colour molecular graphics are often used on chemistry journal covers artistically.[3]
History edit
Prior to the use of computer graphics in representing molecular structure, Robert Corey and Linus Pauling developed a system for representing atoms or groups of atoms from hard wood on a scale of 1 inch = 1 angstrom connected by a clamping device to maintain the molecular configuration.[4] These early models also established the CPK coloring scheme that is still used today to differentiate the different types of atoms in molecular models (e.g. carbon = black, oxygen = red, nitrogen = blue, etc). This early model was improved upon in 1966 by W.L. Koltun and are now known as Corey-Pauling-Koltun (CPK) models.[5]
The earliest efforts to produce models of molecular structure was done by Project MAC using wire-frame models displayed on a cathode ray tube in the mid 1960s. In 1965, Carroll Johnson distributed the Oak Ridge thermal ellipsoid plot (ORTEP) that visualized molecules as a ball-and-stick model with lines representing the bonds between atoms and ellipsoids to represent the probability of thermal motion.[6] Thermal ellipsoid plots quickly became the de facto standard used in the display of X-ray crystallography data, and are still in wide use today.[6] The first practical use of molecular graphics was a simple display of the protein myoglobin using a wireframe representation in 1966 by Cyrus Levinthal and Robert Langridge working at Project MAC.[7]
Among the milestones in high-performance molecular graphics was the work of Nelson Max in "realistic" rendering of macromolecules using reflecting spheres.

Initially much of the technology concentrated on high-performance 3D graphics.[8] During the 1970s, methods for displaying 3D graphics using cathode ray tubes were developed using continuous tone computer graphics in combination with electro-optic shutter viewing devices.[9] The first devices used an active shutter 3D system, generating different perspective views for the left and right channel to provide the illusion of three-dimensional viewing. Stereoscopic viewing glasses were designed using lead lanthanum zirconate titanate (PLZT) ceramics as electronically-controlled shutter elements.[10] Active 3D glasses require batteries and work in concert with the display to actively change the presentation by the lenses to the wearer's eyes. Many modern 3D glasses use a passive, polarized 3D system that enables the wearer to visualize 3D effects based on their own perception. Passive 3D glasses are more common today since they are less expensive.[11]
The requirements of macromolecular crystallography also drove molecular graphics because the traditional techniques of physical model-building could not scale. The first two protein structures solved by molecular graphics without the aid of the Richards' Box were built with Stan Swanson's program FIT on the Vector General graphics display in the laboratory of Edgar Meyer at Texas A&M University: First Marge Legg in Al Cotton's lab at A&M solved a second, higher-resolution structure of staph. nuclease (1975) and then Jim Hogle solved the structure of monoclinic lysozyme in 1976. A full year passed before other graphics systems were used to replace the Richards' Box for modelling into density in 3-D. Alwyn Jones' FRODO program (and later "O") were developed to overlay the molecular electron density determined from X-ray crystallography and the hypothetical molecular structure.
Timeline edit
| Developer(s) | Approximate date | Technology | Comments |
|---|---|---|---|
| Crystallographers | < 1960 | Hand-drawn | Crystal structures, with hidden atom and bond removal. Often clinographic projections. |
| Johnson, Motherwell | c. 1970 | Pen plotter | ORTEP, PLUTO. Very widely deployed for publishing crystal structures. |
| Cyrus Levinthal, Bob Langridge, Ward, Stots[12] | 1966 | Project MAC display system, two-degree of freedom, spring-return velocity joystick for rotating the image. | First protein display on screen. System for interactively building protein structures. |
| Barry[13] | 1969 | LINC 300 computer with a dual trace oscilloscope display. | Interactive molecular structure viewing system. Early examples of dynamic rotation, intensity depth·cueing, and side-by-side stereo. Early use of the small angle approximations (a = sin a, 1 = cos a) to speed up graphical rotation calculations. |
| Ortony | 1971 | Designed a stereo viewer (British patent appl. 13844/70) for molecular computer graphics. | Horizontal two-way (half-silvered) mirror combines images drawn on the upper and lower halves of a CRT. Crossed polarizers isolate the images to each eye. |
| Ortony[14] | 1971 | Light pen, knob. | Interactive molecular structure viewing system. Select bond by turning another knob until desired bond lights up in sequence, a technique later used on the MMS-4 system below, or by picking with the light pen. Points in space are specified with a 3-D ”bug" under dynamic control. |
| Barry, Graesser, Marshall[15] | 1971 | CHEMAST: LINC 300 computer driving an oscilloscope. Two-axis joystick, similar to one used later by GRIP-75 (below). | Interactive molecular structure viewing system. Structures dynamically rotated using the joystick. |
| Tountas and Katz[16] | 1971 | Adage AGT/50 display | Interactive molecular structure viewing system. Mathematics of nested rotation and for laboratory-space rotation. |
| Perkins, Piper, Tattam, White[17] | 1971 | Honeywell DDP 516 computer, EAL TR48 analog computer, Lanelec oscilloscope, 7 linear potentiometers. Stereo. | Interactive molecular structure viewing system. |
| Wright[18][19][20] | 1972 | GRIP-71 at UNC-CH: IBM System/360 Model 40 time-shared computer, IBM 2250 display, buttons, light pen, keyboard. | Discrete manipulation and energy relaxation of protein structures. Program code became the foundation of the GRIP-75 system below. |
| Barry and North[21] | 1972 | University of Oxford: Ferranti Argus 500 computer, Ferranti model 30 display, keyboard, track ball, one knob. Stereo. | Prototype large-molecule crystallographic structure solution system. Track ball rotates a bond, knob brightens the molecule vs. electron density map. |
| North, Ford, Watson | Early 1970s | University of Leeds: DEC PDP·11/40 computer, Hewlett-Packard display. 16 knobs, keyboard, spring-return joystick. Stereo. | Prototype large-molecule crystallographic structure solution system. Six knobs rotate and translate a small molecule. |
| Barry, Bosshard, Ellis, Marshall, Fritch, Jacobi | 1974 | MMS-4:[22][23] Washington University in St. Louis, LINC 300 computer and an LDS-1 / LINC 300 display, custom display modules. Rotation joystick, knobs. Stereo. | Prototype large-molecule crystallographic structure solution system. Select bond to rotate by turning another knob until desired bond lights up in sequence. |
| Cohen and Feldmann[24] | 1974 | DEC PDP-10 computer, Adage display, push buttons, keyboard, knobs | Prototype large-molecule crystallographic structure solution system. |
| Stellman[25] | 1975 | Princeton University: PDP-10 computer, LDS-1 display, knobs | Prototype large-molecule crystallographic structure solution system. Electron density map not shown; instead an "H Factor" figure of merit is updated as the molecular structure is manipulated. |
| Collins, Cotton, Hazen, Meyer, Morimoto | 1975 | CRYSNET,[26] Texas A&M Univ. DEC PDP-11/40 computer, Vector General Series 3 display, knobs, keyboard. Stereo. | Prototype large-molecule crystallographic structure solution system. Variety of viewing modes: rocking, spinning, and several stereo display modes. |
| Cornelius and Kraut | 1976 (approx.) | University of California at San Diego: DEC PDP-11/40 emulator (CalData 135), Evans and Sutherland Picture System display, keyboard, 6 knobs. Stereo. | Prototype large-molecule crystallographic structure solution system. |
| (Yale Univ.) | 1976 (approx.) | PIGS: DEC PDP-11/70 computer, Evans and Sutherland Picture System 2 display, data tablet, knobs. | Prototype large-molecule crystallographic structure solution system. The tablet was used for most interactions. |
| Feldmann and Porter | 1976 | NIH: DEC PDP—11/70 computer. Evans and Sutherland Picture System 2 display, knobs. Stereo. | Interactive molecular structure viewing system. Intended to display interactively molecular data from the AMSOM – Atlas of Macromolecular Structure on Microfiche.[27] |
| Rosenberger et al. | 1976 | MMS-X:[28] Washington University in St. Louis, TI 980B computer, Hewlett-Packard 1321A display, Beehive video terminal, custom display modules, pair of 3-D spring-return joysticks, knobs. | Prototype (and later successful) large-molecule crystallographic structure solution system. Successor to the MMS-4 system above. The 3-D spring-return joysticks either translate and rotate the molecular structure for viewing or a molecular substructure for fitting, mode controlled by a toggle switch. |
| Britton, Lipscomb, Pique, Wright, Brooks | 1977 | GRIP-75[20][29][30][31][32] at UNC-CH: Time-shared IBM System/360 Model 75 computer, DEC PDP 11/45 computer, Vector General Series 3 display, 3-D movement box from A.M. Noll and 3-D spring return joystick for substructure manipulation, Measurement Systems nested joystick, knobs, sliders, buttons, keyboard, light pen. | First large-molecule crystallographic structure solution.[33] |
| Jones | 1978 | FRODO and RING[34][35] Max Planck Inst., Germany, RING: DEC PDP-11/40 and Siemens 4004 computers, Vector General 3404 display, 6 knobs. | Large-molecule crystallographic structure solution. FRODO may have run on a DEC VAX-780 as a follow-on to RING. |
| Diamond | 1978 | Bilder[36] Cambridge, England, DEC PDP-11/50 computer, Evans and Sutherland Picture System display, tablet. | Large-molecule crystallographic structure solution. All input is by data tablet. Molecular structures built on-line with ideal geometry. Later passes stretch bonds with idealization. |
| Langridge, White, Marshall | Late 1970s | Departmental systems (PDP-11, Tektronix displays or DEC-VT11, e.g. MMS-X) | Mixture of commodity computing with early displays. |
| Davies, Hubbard | Mid-1980s | CHEM-X, HYDRA | Laboratory systems with multicolor, raster and vector devices (Sigmex, PS300). |
| Biosym, Tripos, Polygen | Mid-1980s | PS300 and lower cost dumb terminals (VT200, SIGMEX) | Commercial integrated modelling and display packages. |
| Silicon Graphics, Sun | Late 1980s | IRIS GL (UNIX) workstations | Commodity-priced single-user workstations with stereoscopic display. |
| EMBL - WHAT IF | 1989, 2000 | Machine independent | Nearly free, multifunctional, still fully supported, many free servers based on it |
| Sayle, Richardson | 1992, 1993 | RasMol, Kinemage | Platform-independent MG. |
| MDL (van Vliet, Maffett, Adler, Holt) | 1995–1998 | Chime | proprietary C++; free browser plugin for Mac (OS9) and PCs |
| MolSoft | 1997–present | ICM-Browser | proprietary; free download for Windows, Mac, and Linux.[37][38] |
| 1998- | MarvinSketch & MarvinView. MarvinSpace (2005) | proprietary Java applet or stand-alone application. |
Types edit
Ball-and-stick models edit
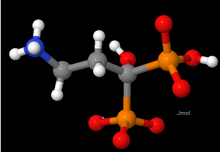
In the ball-and-stick model, atoms are drawn as small sphered connected by rods representing the chemical bonds between them.
Space-filling models edit
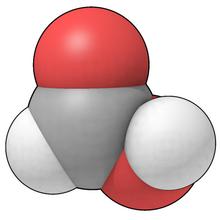
In the space-filling model, atoms are drawn as solid spheres to suggest the space they occupy, in proportion to their van der Waals radii. Atoms that share a bond overlap with each other.
Surfaces edit
In some models, the surface of the molecule is approximated and shaded to represent a physical property of the molecule, such as electronic charge density.[39][40]
Ribbon diagrams edit
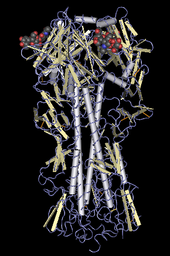
Ribbon diagrams are schematic representations of protein structure and are one of the most common methods of protein depiction used today. The ribbon shows the overall path and organization of the protein backbone in 3D, and serves as a visual framework on which to hang details of the full atomic structure, such as the balls for the oxygen atoms bound to the active site of myoglobin in the adjacent image. Ribbon diagrams are generated by interpolating a smooth curve through the polypeptide backbone. α-helices are shown as coiled ribbons or thick tubes, β-strands as arrows, and non-repetitive coils or loops as lines or thin tubes. The direction of the polypeptide chain is shown locally by the arrows, and may be indicated overall by a colour ramp along the length of the ribbon.[41]
See also edit
References edit
- ^ Dickerson, R.E.; Geis, I. (1969). The structure and action of proteins. Menlo Park, CA: W.A. Benjamin.
- ^ IUPAC, Compendium of Chemical Terminology, 2nd ed. (the "Gold Book") (1997). Online corrected version: (1997) "molecular graphics". doi:10.1351/goldbook.MT06970
- ^ Harrison, Karl; Bowen, Jonathan P.; Bowen, Alice M. (2013). Ng, Kia; Bowen, Jonathan P.; McDaid, Sarah (eds.). "Electronic Visualisation in Chemistry: From Alchemy to Art". EVA London 2013 Conference Proceedings. Electronic Workshops in Computing. British Computer Society. pp. 267–274.
- ^ Corey, Robert B.; Pauling, Linus (1953). "Molecular Models of Amino Acids, Peptides, and Proteins". Review of Scientific Instruments. 24 (8): 621. Bibcode:1953RScI...24..621C. doi:10.1063/1.1770803. Retrieved 13 June 2022.
- ^ Koltun, Walter L. (December 1965). "Precision space-filling atomic models". Biopolymers. 3 (6): 665–679. doi:10.1002/bip.360030606. PMID 4158989. S2CID 38806627.
- ^ a b Atwood, Jerry L.; Barbour, Leonard J. (2003). "Molecular Graphics: From Science to Art". Crystal Growth & Design. 3 (1): 3–8. doi:10.1021/cg020063o. Retrieved 13 June 2022.
- ^ Francoeur, Eric (2002). "Cyrus Levinthal, the Kluge and the origins of interactive molecular graphics". Endeavour. 26 (4): 127–131. doi:10.1016/S0160-9327(02)01468-0. PMID 12535918. Retrieved 13 June 2022.
- ^ Porter TK (August 1978). "Spherical shading". ACM SIGGRAPH Computer Graphics. 12 (3): 282–5. doi:10.1145/965139.639789.
- ^ Ortony, A. (May 1971). "A System for Stereo Viewing". The Computer Journal. 14 (2): 140–4. doi:10.1093/comjnl/14.2.140. Also appears in: Conference on Displays, Institution of Electrical Engineers Conf. Pub. No. 80 (7–10 September 1971), C. Baldwin Ltd., 225-232.
- ^ Roese, John A.; McCleary, Lawrence E. (August 1979). "Stereoscopic Computer Graphics for Simulation and Modeling". ACM SIGGRAPH Computer Graphics. 13 (2): 41–47. doi:10.1145/965103.807423. Retrieved 14 June 2022.
- ^ Wiley, Suzanne S. (12 May 2022). "What Are Passive 3D Glasses?". EasyTechJunkie. Retrieved 14 June 2022.
- ^ Levinthal, C. (June 1966). "Molecular Model-building by Computer". Scientific American. 214 (6): 42–52. Bibcode:1966SciAm.214f..42L. doi:10.1038/scientificamerican0666-42. PMID 5930597.
- ^ Barry, C. D., Ellis, R. A., Graesser, S. M., and Marshall, G. R. 1969. Display and Manipulation in Three Dimensions. Pertinent Comcepts in Computer Graphics, Univ. of Ill. Press, 104-153.
- ^ Ortony, A. 1971b. Interactive Stereographics Conference on Displays, Institution of Electrical Engineers Conf. Pub. No. 80 (7–10 September), C. Baldwin Ltd., 185-193.
- ^ Barry, C. D., Ellis, R. A., Graesser, S. M., and Marshall, G. R. 1971. CHEMAST: A Computer Program for Modeling Molecular Structures. Proc. 1971 IFIP, 1552-1558.
- ^ Tountas, C. and Katz, L. 1971. Interactive Graphics in Molecular Biology. Real·time Three-dimensional Rotations of Images and Image Fragments. Proc. Summer Computer Simulation Conf., 1, 241-247.
- ^ Perkins, W.J.; Piper, E.A.; Tattam, F.G.; White, J.C. (June 1971). "Interactive stereoscopic computer displays for biomedical research". Computers and Biomedical Research. 4 (3): 249–261. doi:10.1016/0010-4809(71)90030-9. PMID 5562569.
- ^ Wright, W. V. 1972a. An Interactive Computer Graphic System for Molecular Studies. PhD Dissertation, University of North Carolina, Chapel Hill, North Carolina.
- ^ Wright, W.V. (October 1972). "The two-dimensional interface of an interactive system for molecular studies". ACM SIGPLAN Notices. 7 (10): 76–85. doi:10.1145/942576.807017.
- ^ a b Brooks FP Jr. The Computer "Scientist" as Toolsmith: Studies in Interactive Computer Graphics. Proc. IFIP, 625-634 (1977).
- ^ Barry CD, North AC (1972). "The use of a computer-controlled display system in the study of molecular conformations". Cold Spring Harb. Symp. Quant. Biol. 36: 577–84. doi:10.1101/SQB.1972.036.01.072. PMID 4508170.
- ^ Barry CD, Bosshard HE, Ellis RA, Marshall GR (December 1974). "Evolving macromodular molecular modeling system". Fed. Proc. 33 (12): 2368–72. PMID 4435239.
- ^ Fritch, J. M., Ellis, R. A., Jacobi T. H., and Marshall, G. R. 1975. A Macromolecular Graphics System for Protein Structure Research. Computers and Graphics, 1, #2/3 (September), 271-278.
- ^ Cohen, G. H. and Feldmann, R. J. 1974. MAP - An Interactive Graphics Computer Program for the Manipulation and Fitting of Protein Molecules to Electron Density Maps. Am. Crystallography. Assoc. Spring 23, (Abstr.).
- ^ Stellman, S.D. (September 1975). "Application of three-dimensional interactive graphics in X-ray crystallographic analysis". Computers & Graphics. 1 (2–3): 279–288. doi:10.1016/0097-8493(75)90019-9.
- ^ Collins DM, Cotton FA, Hazen EE, Meyer EF, Morimoto CN (December 1975). "Protein crystal structures: quicker, cheaper approaches". Science. 190 (4219): 1047–53. Bibcode:1975Sci...190.1047C. doi:10.1126/science.1188383. PMID 1188383. S2CID 44583219.
- ^ Feldmann, R. J. 1976. AMSOM – Atlas of Macromolecular Structure on Microfiche.. Maryland: Tracor Jitco Inc.
- ^ Rosenberger, F. U., et al. 1976. Extracts from 1976 NIH Annual Report. Technical Memorandum No. 230, Computer Systems Laboratory, Washington University in St. Louis, Missouri.
- ^ Lipscomb, JS. Three-dimensional cues for a molecular computer graphics system. PhD Dissertation, University of North Carolina at Chapel Hill, North Carolina. (1981)
- ^ Britton E, Lipscomb JS, Pique ME, Wright, WV, Brooks FP Jr, Pique ME. The GRIP-75 Man-machine Interface. ACM SIGGRAPH Video Review, (4), (Aug. 1981).
- ^ Britton, E. G. 1977. A Methodology for the Ergonomic Design of Interactive Computer Graphics Systems, and its Application to Crystallography. PhD Dissertation, University of North Carolina, Chapel Hill, North Carolina.
- ^ Pique, M. E. 1980. Nested Dynamic Rotations for Computer Graphics. M. S. Thesis, University of North Carolina, Chapel Hill, North Carolina.
- ^ Tsernoglou D, Petsko GA, Tu AT (April 1977). "Protein sequencing by computer graphics". Biochim. Biophys. Acta. 491 (2): 605–8. doi:10.1016/0005-2795(77)90309-9. PMID 857910.
- ^ Jones, T.A. (August 1978). "A Graphics Model Building and Refinement System for Macromolecules". Journal of Applied Crystallography. 11 (4): 268–272. doi:10.1107/S0021889878013308.
- ^ Jones, T. A. 1978b. The RING [user manual]. Max-Planck-Institut fur Biochemie, 8033 Martinsried bei Muchen, Germany.
- ^ Diamond, R. 1978. Bilder. A computer graphics program for bipolymers and its application to interpretation of structure of tobacco mosaic virus protein disks at 2-A resolution. Proc. International Union of Pure and Applied Biochemistry: International Symposium on Structure, Conformation, Function, and Evolution. Madras, India, (4 January), Pergamon Press.
- ^ Abagyan R, Lee WH, Raush E, et al. (February 2006). "Disseminating structural genomics data to the public: from a data dump to an animated story". Trends Biochem. Sci. 31 (2): 76–8. doi:10.1016/j.tibs.2005.12.006. PMID 16406633.
- ^ Raush E, Totrov M, Marsden BD, Abagyan R (2009). "A new method for publishing three-dimensional content". PLOS ONE. 4 (10): e7394. Bibcode:2009PLoSO...4.7394R. doi:10.1371/journal.pone.0007394. PMC 2754609. PMID 19841676.
- ^ Wolfson, Haim; Duhovny, Dina; Tsherniak, Aviad. "Molecular Surface Representation". www.cs.tau.ac.il. Retrieved 14 June 2022.
- ^ O'Donnell, TJ. "The Science and Art of Molecular Surfaces". www.cs.uic.edu. Retrieved 14 June 2022.
- ^ Smith, Thomas J. (27 October 2005). "Displaying and Analyzing Atomic Structures on the Macintosh". Danforth Plant Science Center. Archived from the original on 28 March 2002.
External links edit
- Luminary Series Interview with Robert Langridge Interview by Russ Altman and historical slides.
- History of Visualization of Biological Macromolecules by Eric Martz and Eric Francoeur.