


Summary
In chemistry, molecular symmetry describes the symmetry present in molecules and the classification of these molecules according to their symmetry. Molecular symmetry is a fundamental concept in chemistry, as it can be used to predict or explain many of a molecule's chemical properties, such as whether or not it has a dipole moment, as well as its allowed spectroscopic transitions. To do this it is necessary to use group theory. This involves classifying the states of the molecule using the irreducible representations from the character table of the symmetry group of the molecule. Symmetry is useful in the study of molecular orbitals, with applications to the Hückel method, to ligand field theory, and to the Woodward-Hoffmann rules. Many university level textbooks on physical chemistry, quantum chemistry, spectroscopy and inorganic chemistry discuss symmetry.[1][2][3][4][5][6] Another framework on a larger scale is the use of crystal systems to describe crystallographic symmetry in bulk materials.
There are many techniques for determining the symmetry of a given molecule, including X-ray crystallography and various forms of spectroscopy. Spectroscopic notation is based on symmetry considerations.
Point group symmetry concepts edit
| Rotational axis (Cn) |
Improper rotational elements (Sn) | ||
|---|---|---|---|
| Chiral no Sn |
Achiral mirror plane S1 = σ |
Achiral inversion centre S2 = i | |
| C1 |  |
 |

|
| C2 |  |
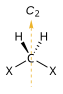 |

|
Elements edit
The point group symmetry of a molecule is defined by the presence or absence of 5 types of symmetry element.
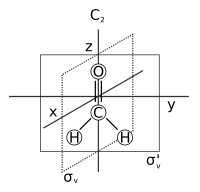
- Symmetry axis: an axis around which a rotation by results in a molecule indistinguishable from the original. This is also called an n-fold rotational axis and abbreviated Cn. Examples are the C2 axis in water and the C3 axis in ammonia. A molecule can have more than one symmetry axis; the one with the highest n is called the principal axis, and by convention is aligned with the z-axis in a Cartesian coordinate system.
- Plane of symmetry: a plane of reflection through which an identical copy of the original molecule is generated. This is also called a mirror plane and abbreviated σ (sigma = Greek "s", from the German 'Spiegel' meaning mirror).[7] Water has two of them: one in the plane of the molecule itself and one perpendicular to it. A symmetry plane parallel with the principal axis is dubbed vertical (σv) and one perpendicular to it horizontal (σh). A third type of symmetry plane exists: If a vertical symmetry plane additionally bisects the angle between two 2-fold rotation axes perpendicular to the principal axis, the plane is dubbed dihedral (σd). A symmetry plane can also be identified by its Cartesian orientation, e.g., (xz) or (yz).
- Center of symmetry or inversion center, abbreviated i. A molecule has a center of symmetry when, for any atom in the molecule, an identical atom exists diametrically opposite this center an equal distance from it. In other words, a molecule has a center of symmetry when the points (x,y,z) and (−x,−y,−z) are identical. For example, if there is an oxygen atom in some point (x,y,z), then there is an oxygen atom in the point (−x,−y,−z). There may or may not be an atom at the inversion center itself. Examples are xenon tetrafluoride (a square planar molecule), where the inversion center is at the Xe atom, and benzene (C
6H
6) where the inversion center is at the center of the ring. - Rotation-reflection axis: an axis around which a rotation by , followed by a reflection in a plane perpendicular to it, leaves the molecule unchanged. Also called an n-fold improper rotation axis, it is abbreviated Sn. Examples are present in tetrahedral silicon tetrafluoride, with three S4 axes, and the staggered conformation of ethane with one S6 axis. An S1 axis corresponds to a mirror plane σ and an S2 axis is an inversion center i. A molecule which has no Sn axis for any value of n is a chiral molecule.
- Identity, abbreviated to E, from the German 'Einheit' meaning unity.[8] This symmetry element simply consists of no change: every molecule has this symmetry element, which is equivalent to a C1 proper rotation. It must be included in the list of symmetry elements so that they form a mathematical group, whose definition requires inclusion of the identity element. It is so called because it is analogous to multiplying by one (unity).[9]


Operations edit

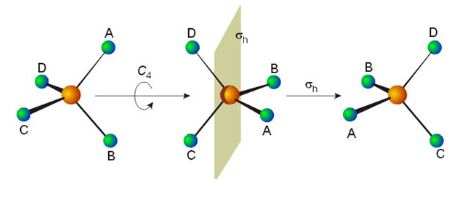
The five symmetry elements have associated with them five types of symmetry operation, which leave the geometry of the molecule indistinguishable from the starting geometry. They are sometimes distinguished from symmetry elements by a caret or circumflex. Thus, Ĉn is the rotation of a molecule around an axis and Ê is the identity operation. A symmetry element can have more than one symmetry operation associated with it. For example, the C4 axis of the square xenon tetrafluoride (XeF4) molecule is associated with two Ĉ4 rotations in opposite directions (90° and 270°), a Ĉ2 rotation (180°) and Ĉ1 (0° or 360°). Because Ĉ1 is equivalent to Ê, Ŝ1 to σ and Ŝ2 to î, all symmetry operations can be classified as either proper or improper rotations.
For linear molecules, either clockwise or counterclockwise rotation about the molecular axis by any angle Φ is a symmetry operation.

Symmetry groups edit
Groups edit
The symmetry operations of a molecule (or other object) form a group. In mathematics, a group is a set with a binary operation that satisfies the four properties listed below.
In a symmetry group, the group elements are the symmetry operations (not the symmetry elements), and the binary combination consists of applying first one symmetry operation and then the other. An example is the sequence of a C4 rotation about the z-axis and a reflection in the xy-plane, denoted σ(xy)C4. By convention the order of operations is from right to left.
A symmetry group obeys the defining properties of any group.
- closure property:
For every pair of elements x and y in G, the product x*y is also in G.( in symbols, for every two elements x, y ∈ G, x*y is also in G ).This means that the group is closed so that combining two elements produces no new elements. Symmetry operations have this property because a sequence of two operations will produce a third state indistinguishable from the second and therefore from the first, so that the net effect on the molecule is still a symmetry operation. This may be illustrated by means of a table. For example, with the point group C3, there are three symmetry operations: rotation by 120°, C3, rotation by 240°, C32 and rotation by 360°, which is equivalent to identity, E.
C2v point group multiplication tab Point group C3 Multiplication table E C3 C32 E E C3 C32 C3 C3 C32 E C32 C32 E C3
- This table also illustrates the following properties
- Associative property:
For every x and y and z in G, both (x*y)*z and x*(y*z) result with the same element in G.( in symbols, (x*y)*z = x*(y*z ) for every x, y, and z ∈ G)
- existence of identity property:
There must be an element ( say e ) in G such that product any element of G with e make no change to the element.( in symbols, x*e = e*x = x for every x ∈ G )
- existence of inverse element:
For each element x in G, there must be an element y in G such that product of x and y is the identity element e.( in symbols, for each x ∈ G there is a y ∈ G such that x*y = y*x = e for every x ∈ G )

The order of a group is the number of elements in the group. For groups of small orders, the group properties can be easily verified by considering its composition table, a table whose rows and columns correspond to elements of the group and whose entries correspond to their products.
Point groups and permutation-inversion groups edit

The successive application (or composition) of one or more symmetry operations of a molecule has an effect equivalent to that of some single symmetry operation of the molecule. For example, a C2 rotation followed by a σv reflection is seen to be a σv' symmetry operation: σv*C2 = σv'. ("Operation A followed by B to form C" is written BA = C).[9] Moreover, the set of all symmetry operations (including this composition operation) obeys all the properties of a group, given above. So (S,*) is a group, where S is the set of all symmetry operations of some molecule, and * denotes the composition (repeated application) of symmetry operations.
This group is called the point group of that molecule, because the set of symmetry operations leave at least one point fixed (though for some symmetries an entire axis or an entire plane remains fixed). In other words, a point group is a group that summarises all symmetry operations that all molecules in that category have.[9] The symmetry of a crystal, by contrast, is described by a space group of symmetry operations, which includes translations in space.
One can determine the symmetry operations of the point group for a particular molecule by considering the geometrical symmetry of its molecular model. However, when one uses a point group to classify molecular states, the operations in it are not to be interpreted in the same way. Instead the operations are interpreted as rotating and/or reflecting the vibronic (vibration-electronic) coordinates[10] and these operations commute with the vibronic Hamiltonian. They are "symmetry operations" for that vibronic Hamiltonian. The point group is used to classify by symmetry the vibronic eigenstates of a rigid molecule. The symmetry classification of the rotational levels, the eigenstates of the full (rotation-vibration-electronic) Hamiltonian, requires the use of the appropriate permutation-inversion group as introduced by Longuet-Higgins.[11] Point groups describe the geometrical symmetry of a molecule whereas permutation-inversion groups describe the energy-invariant symmetry.
Examples of point groups edit
Assigning each molecule a point group classifies molecules into categories with similar symmetry properties. For example, PCl3, POF3, XeO3, and NH3 all share identical symmetry operations.[12] They all can undergo the identity operation E, two different C3 rotation operations, and three different σv plane reflections without altering their identities, so they are placed in one point group, C3v, with order 6.[9] Similarly, water (H2O) and hydrogen sulfide (H2S) also share identical symmetry operations. They both undergo the identity operation E, one C2 rotation, and two σv reflections without altering their identities, so they are both placed in one point group, C2v, with order 4.[13] This classification system helps scientists to study molecules more efficiently, since chemically related molecules in the same point group tend to exhibit similar bonding schemes, molecular bonding diagrams, and spectroscopic properties.[9] Point group symmetry describes the symmetry of a molecule when fixed at its equilibrium configuration in a particular electronic state. It does not allow for tunneling between minima nor for the change in shape that can come about from the centrifugal distortion effects of molecular rotation.
Common point groups edit
The following table lists many of the point groups applicable to molecules, labelled using the Schoenflies notation, which is common in chemistry and molecular spectroscopy. The descriptions include common shapes of molecules, which can be explained by the VSEPR model. In each row, the descriptions and examples have no higher symmetries, meaning that the named point group captures all of the point symmetries.
| Point group | Symmetry operations[14] | Simple description of typical geometry | Example 1 | Example 2 | Example 3 |
|---|---|---|---|---|---|
| C1 | E | no symmetry, chiral |  bromochlorofluoromethane (both enantiomers shown) |
 lysergic acid |
 L-leucine and most other α-amino acids except glycine |
| Cs | E σh | mirror plane |  thionyl chloride |
hypochlorous acid |
 chloroiodomethane |
| Ci | E i | inversion center |  meso-tartaric acid |
 mucic acid (meso-galactaric acid) |
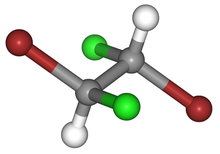 |
| C∞v | E 2C∞Φ ∞σv | linear |  hydrogen fluoride (and all other heteronuclear diatomic molecules) |
 nitrous oxide (dinitrogen monoxide) |
 hydrocyanic acid (hydrogen cyanide) |
| D∞h | E 2C∞Φ ∞σi i 2S∞Φ ∞C2 | linear with inversion center | oxygen (and all other homonuclear diatomic molecules) |
 carbon dioxide |
 acetylene (ethyne) |
| C2 | E C2 | "open book geometry", chiral |  hydrogen peroxide |
 hydrazine |
 tetrahydrofuran (twist conformation) |
| C3 | E C3 C32 | propeller, chiral |  triphenylphosphine |
 triethylamine |
 phosphoric acid |
| C2h | E C2 i σh | planar with inversion center, no vertical plane | trans-1,2-dichloroethylene |
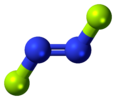 trans-dinitrogen difluoride |
trans-azobenzene |
| C2v | E C2 σv(xz) σv'(yz) | angular (H2O) or see-saw (SF4) |  water |
 sulfur tetrafluoride |
 Dichloromethane |
| C3h | E C3 C32 σh S3 S35 | propeller |  boric acid |
 phloroglucinol (1,3,5-trihydroxybenzene) |
 |
| C3v | E 2C3 3σv | trigonal pyramidal | 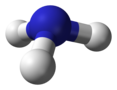 ammonia (if pyramidal inversion is neglected) |
 phosphorus oxychloride |
 cobalt tetracarbonyl hydride, HCo(CO)4 |
| C4v | E 2C4 C2 2σv 2σd | square pyramidal |  xenon oxytetrafluoride |
 pentaborane(9), B5H9 |
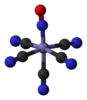 nitroprusside anion [Fe(CN)5(NO)]2− |
| C5 | E 2C5 2C52 | five-fold rotational symmetry |  C-reactive protein |
 |
 |
| C5v | E 2C5 2C52 5σv | 'milking stool' complex | 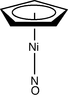 Cyclopentadienyl nickel nitrosyl (CpNiNO) |
 corannulene |
 |
| D2 | E C2(x) C2(y) C2(z) | twist, chiral |  biphenyl (skew conformation) |
 twistane (C10H16) |
|
| D3 | E C3(z) 3C2 | triple helix, chiral |  Tris(ethylenediamine)cobalt(III) cation |
tris(oxalato)iron(III) anion |
 |
| D2h | E C2(z) C2(y) C2(x) i σ(xy) σ(xz) σ(yz) | planar with inversion center, vertical plane |  ethylene |
 pyrazine |
 diborane |
| D3h | E 2C3 3C2 σh 2S3 3σv | trigonal planar or trigonal bipyramidal |  boron trifluoride |
phosphorus pentachloride |
 cyclopropane |
| D4h | E 2C4 C2 2C2' 2C2" i 2S4 σh 2σv 2σd | square planar |  xenon tetrafluoride |
 octachlorodimolybdate(II) anion |
Trans-[CoIII(NH3)4Cl2]+ (excluding H atoms) |
| D5h | E 2C5 2C52 5C2 σh 2S5 2S53 5σv | pentagonal |  cyclopentadienyl anion |
 ruthenocene |
 C70 |
| D6h | E 2C6 2C3 C2 3C2' 3C2‘’ i 2S3 2S6 σh 3σd 3σv | hexagonal |  benzene |
 bis(benzene)chromium |
 coronene (C24H12) |
| D7h | E C7 S7 7C2 σh 7σv | heptagonal |  tropylium (C7H7+) cation |
 |
|
| D8h | E C8 C4 C2 S8 i 8C2 σh 4σv 4σd | octagonal |  cyclooctatetraenide (C8H82−) anion |
 uranocene |
 |
| D2d | E 2S4 C2 2C2' 2σd | 90° twist |  allene |
 tetrasulfur tetranitride |
 diborane(4) (excited state) |
| D3d | E 2C3 3C2 i 2S6 3σd | 60° twist |  ethane (staggered rotamer) |
 dicobalt octacarbonyl (non-bridged isomer) |
 cyclohexane chair conformation |
| D4d | E 2S8 2C4 2S83 C2 4C2' 4σd | 45° twist |  sulfur (crown conformation of S8) |
 dimanganese decacarbonyl (staggered rotamer) |
 octafluoroxenate ion (idealized geometry) |
| D5d | E 2C5 2C52 5C2 i 2S103 2S10 5σd | 36° twist | ferrocene (staggered rotamer) |
 |
 |
| S4 | E 2S4 C2 |  1,2,3,4-tetrafluorospiropentane (meso isomer)[15] |
 |
 | |
| Td | E 8C3 3C2 6S4 6σd | tetrahedral |  methane |
 phosphorus pentoxide |
 adamantane |
| Th | E 4C3 4C32 i 3C2 4S6 4S65 3σh | pyritohedron |  |
 |
 |
| Oh | E 8C3 6C2 6C4 3C2 i 6S4 8S6 3σh 6σd | octahedral or cubic |  sulfur hexafluoride |
 molybdenum hexacarbonyl |
 cubane |
| I | E 12C5 12C52 20C3 15C2 | chiral icosahedral or dodecahedral |  Rhinovirus |
 |
 |
| Ih | E 12C5 12C52 20C3 15C2 i 12S10 12S103 20S6 15σ | icosahedral or dodecahedral |  Buckminsterfullerene |
 dodecaborate anion |
 dodecahedrane |
Representations edit
A set of matrices that multiply together in a way that mimics the multiplication table of the elements of a group is called a representation of the group. For example, for the C2v point group, the following three matrices are part of a representation of the group:

Although an infinite number of such representations exist, the irreducible representations (or "irreps") of the group are all that are needed as all other representations of the group can be described as a direct sum of the irreducible representations. Also, the irreducibile representations are those matrix representations in which the matrices are in their most diagonal form possible.

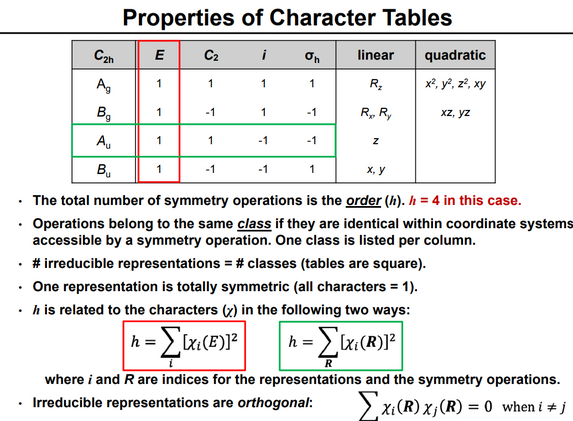
Character tables edit
For any group, its character table gives a tabulation (for the classes of the group) of the characters (the sum of the diagonal elements) of the matrices of all the irreducible representations of the group. As the number of irreducible representations equals the number of classes, the character table is square.
The representations are labeled according to a set of conventions:
- A, when rotation around the principal axis is symmetrical
- B, when rotation around the principal axis is asymmetrical
- E and T are doubly and triply degenerate representations, respectively
- when the point group has an inversion center, the subscript g (German: gerade or even) signals no change in sign, and the subscript u (ungerade or uneven) a change in sign, with respect to inversion.
- with point groups C∞v and D∞h the symbols are borrowed from angular momentum description: Σ, Π, Δ.
The tables also capture information about how the Cartesian basis vectors, rotations about them, and quadratic functions of them transform by the symmetry operations of the group, by noting which irreducible representation transforms in the same way. These indications are conventionally on the righthand side of the tables. This information is useful because chemically important orbitals (in particular p and d orbitals) have the same symmetries as these entities.
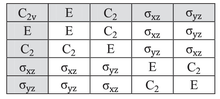
The character table for the C2v symmetry point group is given below:
| C2v | E | C2 | σv(xz) | σv'(yz) | ||
|---|---|---|---|---|---|---|
| A1 | 1 | 1 | 1 | 1 | z | x2, y2, z2 |
| A2 | 1 | 1 | −1 | −1 | Rz | xy |
| B1 | 1 | −1 | 1 | −1 | x, Ry | xz |
| B2 | 1 | −1 | −1 | 1 | y, Rx | yz |
Consider the example of water (H2O), which has the C2v symmetry described above. The 2px orbital of oxygen has B1 symmetry as in the fourth row of the character table above, with x in the sixth column). It is oriented perpendicular to the plane of the molecule and switches sign with a C2 and a σv'(yz) operation, but remains unchanged with the other two operations (obviously, the character for the identity operation is always +1). This orbital's character set is thus {1, −1, 1, −1}, corresponding to the B1 irreducible representation. Likewise, the 2pz orbital is seen to have the symmetry of the A1 irreducible representation (i.e.: none of the symmetry operations change it), 2py B2, and the 3dxy orbital A2. These assignments and others are noted in the rightmost two columns of the table.
Historical background edit
Hans Bethe used characters of point group operations in his study of ligand field theory in 1929, and Eugene Wigner used group theory to explain the selection rules of atomic spectroscopy.[16] The first character tables were compiled by László Tisza (1933), in connection to vibrational spectra. Robert Mulliken was the first to publish character tables in English (1933), and E. Bright Wilson used them in 1934 to predict the symmetry of vibrational normal modes.[17] The complete set of 32 crystallographic point groups was published in 1936 by Rosenthal and Murphy.[18]
Symmetry of vibrational modes edit
Each normal mode of molecular vibration has a symmetry which forms a basis for one irreducible representation of the molecular symmetry group.[19] For example, the water molecule has three normal modes of vibration: symmetric stretch in which the two O-H bond lengths vary in phase with each other, asymmetric stretch in which they vary out of phase, and bending in which the bond angle varies. The molecular symmetry of water is C2v with four irreducible representations A1, A2, B1 and B2. The symmetric stretching and the bending modes have symmetry A1, while the asymmetric mode has symmetry B2. The overall symmetry of the three vibrational modes is therefore Γvib = 2A1 + B2.[19][20]
Vibrational modes of ammonia edit
The molecular symmetry of ammonia is C3v. The number of vibrational modes can be found by using the formula 3N-6 for non-linear molecule. So it has six vibrational modes. It has E, C3 and σv symmetry operations.[7] The motion of the nitrogen atom and the three hydrogen atoms in relation to one another give birth to these modes. All three hydrogen atoms travel symmetrically along the molecule's axis, either in the direction of the nitrogen atom or away from it. This mode is known as symmetric stretch (v₁). The symmetry in the N-H bond stretching is reflected in this stretching motion. Of the three vibrational modes, this one has the highest frequency.[21]

In Bending (ν₂) configuration, the nitrogen atom stays still while the three hydrogen atoms move in different directions from one another. Changes in the bond angles result from this action, which includes the molecule bending out of plane. Because of how the hydrogen atoms move like an umbrella, this mode is often referred to as the "umbrella mode".[23] Asymmetric Stretch (ν₃) is also named as asymmetric stretching mode. While one atom approaches the nitrogen atom, two hydrogen atoms move apart in this mode.
Ammonia has four atoms. Each atom is associated with three vectors. NH3 has three irreducible representation A1, A2 and E. Total modes of vibration includes the vibrational, rotational and translational modes.
Total modes = 3A1 + A2 + 4E
Rotational modes = A2 + E
Translational mode = A1 + E
Vibrational mode = Total mode - Rotational mode - Translational mode = 3A1 + A2 + 4E - A2 - E - A1 - E = 2A1 + 2E
More examples edit
- W(CO)6 has octahedral geometry. The irreducible representation for the C-O stretching vibration is A1g + Eg + T1u . Out of these, only T1u is IR active.
- B2H6 (diborane) has D2h molecular symmetry. The terminal B-H stretching vibrations which are active in IR are B2u and B3u.

- Fac-Mo(CO)3(CH3CH2CN)3, has C3v geometry. The irreducible representation for the C-O stretching vibration is A1 + E. Both of which are IR active.
Symmetry of molecular orbitals edit
Each molecular orbital also has the symmetry of one irreducible representation. For example, ethylene (C2H4) has symmetry group D2h, and its highest occupied molecular orbital (HOMO) is the bonding pi orbital which forms a basis for its irreducible representation B1u.[24]
Molecular rotation and molecular nonrigidity edit
As discussed above in the section Point groups and permutation-inversion groups, point groups are useful for classifying the vibrational and electronic states of rigid molecules (sometimes called semi-rigid molecules) which undergo only small oscillations about a single equilibrium geometry. Longuet-Higgins introduced a more general type of symmetry group[11] suitable not only for classifying the vibrational and electronic states of rigid molecules but also for classifying their rotational and nuclear spin states. Further, such groups can be used to classify the states of non-rigid (or fluxional) molecules that tunnel between equivalent geometries (called versions[25]) and to allow for the distorting effects of molecular rotation. These groups are known as permutation-inversion groups, because the symmetry operations in them are energetically feasible permutations of identical nuclei, or inversion with respect to the center of mass (the parity operation), or a combination of the two.
For example, ethane (C2H6) has three equivalent staggered conformations. Tunneling between the conformations occurs at ordinary temperatures by internal rotation of one methyl group relative to the other. This is not a rotation of the entire molecule about the C3 axis. Although each conformation has D3d symmetry, as in the table above, description of the internal rotation and associated quantum states and energy levels requires the more complete permutation-inversion group G36.[26]
Similarly, ammonia (NH3) has two equivalent pyramidal (C3v) conformations which are interconverted by the process known as nitrogen inversion. This is not the point group inversion operation i used for centrosymmetric rigid molecules (i.e., the inversion of vibrational displacements and electronic coordinates in the nuclear center of mass) since NH3 has no inversion center and is not centrosymmetric. Rather, it is the inversion of the nuclear and electronic coordinates in the molecular center of mass (sometimes called the parity operation), which happens to be energetically feasible for this molecule. The appropriate permutation-inversion group to be used in this situation is D3h(M)[27] which is isomorphic with the point group D3h.
Additionally, as examples, the methane (CH4) and H3+ molecules have highly symmetric equilibrium structures with Td and D3h point group symmetries respectively; they lack permanent electric dipole moments but they do have very weak pure rotation spectra because of rotational centrifugal distortion.[28][29] The permutation-inversion groups required for the complete study of CH4 and H3+ are Td(M)[30] and D3h(M), respectively.
In its ground (N) electronic state the ethylene molecule C2H4 has D2h point group symmetry whereas in the excited (V) state it has D2d symmetry. To treat these two states together it is necessary to allow torsion and to use the double group of the permutation-inversion group G16.[31]
A second and less general approach to the symmetry of nonrigid molecules is due to Altmann.[32][33] In this approach the symmetry groups are known as Schrödinger supergroups and consist of two types of operations (and their combinations): (1) the geometric symmetry operations (rotations, reflections, inversions) of rigid molecules, and (2) isodynamic operations, which take a nonrigid molecule into an energetically equivalent form by a physically reasonable process such as rotation about a single bond (as in ethane) or a molecular inversion (as in ammonia).[33]
See also edit
- Parity (physics) § Molecules
- Irreducible representation § Applications in theoretical physics and chemistry
- Woodward-Hoffmann rules § Correlation diagrams
- Hapticity § Hapticity and fluxionality
- Character table
- Crystallographic point group
- Point groups in three dimensions
- Symmetry of diatomic molecules
- Symmetry in quantum mechanics
References edit
- ^ Quantum Chemistry, 3rd ed. John P. Lowe, Kirk Peterson ISBN 0-12-457551-X
- ^ Physical Chemistry: A Molecular Approach by Donald A. McQuarrie, John D. Simon ISBN 0-935702-99-7
- ^ The chemical bond, 2nd ed. J.N. Murrell, S.F.A. Kettle, J.M. Tedder ISBN 0-471-90760-X
- ^ Physical Chemistry, 8th ed. P.W. Atkins and J. de Paula, W.H. Freeman, 2006 ISBN 0-7167-8759-8, chap.12
- ^ G. L. Miessler and D. A. Tarr Inorganic Chemistry, 2nd ed. Pearson, Prentice Hall, 1998 ISBN 0-13-841891-8, chap.4.
- ^ Molecular Symmetry and Spectroscopy, 2nd ed. Philip R. Bunker and Per Jensen, NRC Research Press, Ottawa (1998)[1]ISBN 9780660196282
- ^ a b "Symmetry Operations and Character Tables". University of Exeter. 2001. Retrieved 29 May 2018.
- ^ LEO Ergebnisse für "einheit"
- ^ a b c d e Pfenning, Brian (2015). Principles of Inorganic Chemistry. John Wiley & Sons. ISBN 9781118859025.
- ^ P. R. Bunker and P. Jensen (2005),Fundamentals of Molecular Symmetry (CRC Press)ISBN 0-7503-0941-5 [2] Section 8.3
- ^ a b Longuet-Higgins, H.C. (1963). "The symmetry groups of non-rigid molecules". Molecular Physics. 6 (5): 445–460. Bibcode:1963MolPh...6..445L. doi:10.1080/00268976300100501.
- ^ Pfennig, Brian (30 March 2015). Principles of Inorganic Chemistry. Wiley. p. 191. ISBN 978-1-118-85910-0.
- ^ Miessler, Gary (2004). Inorganic Chemistry. Pearson. ISBN 9780321811059.
- ^ Miessler, Gary L.; Tarr, Donald A. (1999). "Character tables (all except D7h)". Inorganic Chemistry (2nd ed.). Prentice-Hall. pp. 621–630. ISBN 0-13-841891-8.
- ^ Housecroft, C. E.; Sharpe, A. G. (2008). Inorganic Chemistry (3rd ed.). Prentice Hall. pp. 111–112. ISBN 978-0-13-175553-6.
- ^ Group Theory and its application to the quantum mechanics of atomic spectra, E. P. Wigner, Academic Press Inc. (1959)
- ^ Correcting Two Long-Standing Errors in Point Group Symmetry Character Tables Randall B. Shirts J. Chem. Educ. 2007, 84, 1882. Abstract
- ^ Rosenthal, Jenny E.; Murphy, G. M. (1936). "Group Theory and the Vibrations of Polyatomic Molecules". Rev. Mod. Phys. 8 (4): 317–346. Bibcode:1936RvMP....8..317R. doi:10.1103/RevModPhys.8.317.
- ^ a b Harris, Daniel C.; Bertolucci, Michael D. (1978). "3". Symmetry and Spectroscopy. Oxford University Press. pp. 138–142. ISBN 0-19-502001-4.
Each normal mode of vibration will form a basis set for an irreducible representation of the point group of the molecule.
- ^ G. L. Miessler and D. A. Tarr Inorganic Chemistry, 2nd ed. Pearson, Prentice Hall, 1998 ISBN 0-13-841891-8, pp.97-100.
- ^ "Vibrational Modes of Ammonia". www.chem.purdue.edu. Retrieved 2024-04-13.
- ^ Greenwood, N. N.; Earnshaw, A. (1997) Chemistry of the Elements (2nd ed.), Oxford:Butterworth-Heinemann, pp. p. 423 ISBN 0-7506-3365-4
- ^ "Umbrella mode - Big Chemical Encyclopedia". chempedia.info. Retrieved 2024-04-13.
- ^ Harris, Daniel C.; Bertolucci, Michael D. (1978). "4". Symmetry and Spectroscopy. Oxford University Press. p. 278. ISBN 0-19-502001-4.
- ^ Bone, R.G.A.; et al. (1991). "Transition states from molecular symmetry groups:Analysis of non-rigid acetylene trimer". Molecular Physics. 72 (1): 33–73. Bibcode:1991MolPh..72...33B. doi:10.1080/00268979100100021.
- ^ P. R. Bunker (1964). "The Rotation-Torsion Wavefunctions of Molecules that have two Identical Rotors". Mol. Phys. 8: 81. doi:10.1080/00268976400100091.
- ^ P.R. Bunker 'Practically Everything you Ought to know about the Molecular Symmetry Group' in, ‘Vibrational Spectra and Structure, Vol. III’, ed. James R. Durig, Marcel Dekker (1975) ISBN 0824711491
- ^ Watson, J.K.G (1971). "Forbidden rotational spectra of polyatomic molecules". Journal of Molecular Spectroscopy. 40 (3): 546–544. Bibcode:1971JMoSp..40..536W. doi:10.1016/0022-2852(71)90255-4.
- ^ Oldani, M.; et al. (1985). "Pure rotational spectra of methane and methane-d4 in the vibrational ground state observed by microwave Fourier transform spectroscopy". Journal of Molecular Spectroscopy. 110 (1): 93–105. Bibcode:1985JMoSp.110...93O. doi:10.1016/0022-2852(85)90215-2.
- ^ P. R. Bunker; Per Jensen (1999). "Spherical top molecules and the molecular symmetry group". Mol. Phys. 97 (1–2): 255. Bibcode:1999MolPh..97..255B. doi:10.1080/00268979909482827.
- ^ Watson, J.K.G.; et al. (1996). "Theory of odd torsional transitions in the V−N resonance Raman spectrum of ethylene". J Chem Phys. 105 (4): 1348. Bibcode:1996JChPh.105.1348W. doi:10.1063/1.472001.
- ^ Altmann S.L. (1977) Induced Representations in Crystals and Molecules, Academic Press
- ^ a b Flurry, R.L. (1980) Symmetry Groups, Prentice-Hall, ISBN 0-13-880013-8, pp.115-127
External links edit
- Point group symmetry @ Newcastle University
- Molecular symmetry @ Imperial College London
- Molecular Point Group Symmetry Tables
- Character tables for point groups for chemistry
- Molecular Symmetry Online @ The Open University of Israel
- An internet lecture course on molecular symmetry @ Bergische Universitaet
- DECOR – Symmetry @ The Cambridge Crystallographic Data Centre
- Details of the relation between point groups and permutation-inversion groups, by Philip Bunker