


Summary
Organomolybdenum chemistry is the chemistry of chemical compounds with Mo-C bonds. The heavier group 6 elements molybdenum and tungsten form organometallic compounds similar to those in organochromium chemistry but higher oxidation states tend to be more common.[2]

Mo(0) and more reduced states edit
Molybdenum hexacarbonyl is the precursor to many substituted derivatives. It reacts with organolithium reagents to give anionic acyls which can be O-alkylated to give Fischer carbenes.
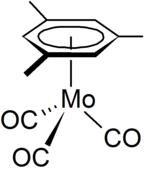
Mo(CO)6 reacts with arenes to give piano-stool complexes such as (mesitylene)molybdenum tricarbonyl. Cycloheptatrienemolybdenum tricarbonyl, which is related to (arene)Mo(CO)3, reacts with trityl salts to give the cycloheptatrienyl complex:[3]
- (C7H8)Mo(CO)3 + (C6H5)3C+ → [(C7H7)Mo(CO)3]+ + (C6H5)3CH

Reduction of Mo(CO)6 gives [Mo(CO)5]2− which is formally Mo(-II).[4]
CO-free Mo(0) compounds tend to be more reducing and kinetically labile than the carbonyl complexes.[5] Examples include bis(benzene)molybdenum (Mo(C6H6)2) and tris(butadiene)molybdenum. Such compounds can be prepared by metal vapor synthesis and reductive routes from molybdenum(V) chloride.[6]
Mo(II) edit
Halogenation of Mo(CO)6 gives Mo(II) carbonyl halides, which are also versatile precursors.[7] One large collection of compounds have the formula (C5R5)Mo(CO)3X, derived from cyclopentadienylmolybdenum tricarbonyl dimer (X = halide, hydride, alkyl).[8]
Treating molybdenum(II) acetate with methyllithium gives Li4[Mo2(CH3)8].
Mo(IV) edit
With the formula of the type Cp2MoX2 molybdocene dichloride (X = Cl) and molybdocene dihydride (X = H) are both known as are ansa metallocene analogues.
Mo(V) and Mo(VI) edit
Mo(CH3)5, Mo(CH3)6, and salts of [Mo(CH3)7]− are known.[5]
Oxo and imide (RN=) ligands are found in several high oxidation state organomolybdenum compounds. The complexes (C5R5)MoO2X are illustrative.[9] Schrock's Mo-based olefin metathesis catalysts feature molybdenum(VI) centers supported by alkoxide, alkylidene, and imido ligands.[10]
Molybdenum neopentylidyne complexes endowed with sterically demanding phenolates or branched fluorinated alkoxides are catalysts for alkyne metathesis.[11] However, preparation of these catalysts is problematic by the standard Schrock procedure. The trisalkoxide species 17 is active at room temperature.[12]
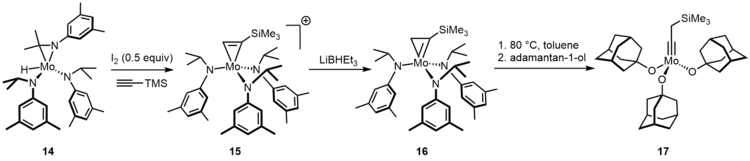
The related complex precursor complex 18 provides even greater opportunities, which is originally designed for the stoichiometric cleavage of dinitrogen.[13][14] In fact, when treating complex 18 with DCM in toluene, the major species formed is a methylidyne complex 19 and a monochloride compound 20.[15] More importantly, the combination of complex 18 and DCM tolerates numerous polar groups. For instance, basic amines and sulfides, which deactivate the more Lewis acidic complex such as Schrock complex. Following by this original discovery, Moore and co-workers tried higher gem-dichlorides RCHCl2 as activating agents to increase the catalyst lifetime.[16] To reconvert the chloride byproduct, they added magnesium in reaction. Moreover, after ligand exchange to an electron deficient ligand such as p-nitrophenol, gave access to a very active catalyst 22, which was effective in many applications, particularly in polymer chemistry and material science.[17] On the other hand, alcoholysis of 21 with a tridentate ligand will lead to longer lifetime and better substrate scope.[18]

Despite the favorable characteristics of such catalysts, complex 18 must be handled with great care. This compound is not only very sensitive to oxidation and hydrolysis, but even reactive enough to cleave molecular nitrogen.
Molybdenum nitride complexes with Ph3SiO ligands are practical and tolerant precatalyst for alkyne metathesis.[19] This result implied that molybdenum alkylidynes endowed with Ph3SiO ligands must be very active. To further increase the feasibility, stability and activity of these catalysts, they came up with an independent route to directly prepare the alkylidynes instead of their nitrile counterparts. By complexation with 1,10-phenanthroline, an air-stable compound 27 can be formed as precatalyst, which can be activated easily by MnCl2 or ZnCl2 in solvents.[20] As shown below, this route is highly scalable and practical.

Organotungsten compounds edit
Tungsten analogues of almost all organoMo compounds are known. Some notable examples include hexamethyltungsten and analogues of Schrock olefin metathesis catalysts.
Many tungsten-based alkyne metathesis catalysts are of the general type [X3W≡CR].[21] Activity is manipulated by the ligands. A typical route to such catalysts entails treatment neopentyl Grignard reagent to tungsten(VI) precursor followed by net alcoholysis of the alkyl ligands.[22] Complex 3 can undergo a ligand exchange with lithium salts to generate Schrock type catalysts (complex 4). Another way to make complex 4 is via cleavage of internal alkyne by W(III) complex, such as 5.[23][24] Complex 2, as well as 3, is unable to metathesize internal alkynes, the related pathway is shown right. In detail, compound 6 (when X is not OR) will react with two equivalent alkynes to form complex 7. Complex 7 will undergo an "associative path" to generate a metallabenzene complex 8. It will decompose to polymerized compounds or a cyclopentadienyl complex with a formally reduced tungsten center. Tungstenocenes, or tungsten-containing metallocenes, may be formed from these cyclopentadienyl complexes.
The formal 12-electron count of the W(VI) center in Schrock catalyst represents an appreciable Lewis acidity, which seriously limits the scope of these catalysts. For example, Schrock catalyst is unable to metathesize substrates containing donor or basic sites such as amines, thio ethers or crown ether segments. Acid-sensitive groups such as acetals can be destroyed. Replacement of tert-butoxide ligands by fluorinated alkoxides increase the Lewis acidic character. To reach a balance, it is proposed that a heteroleptic push/pull environment around the tungsten center will work.(as shown below)[25][26][27][28][29] For example, complex 13 is highly active (with loading 1-2 mol% being sufficient) and compatible with many functional groups.
Applications edit
Mo-based catalysts are useful for olefin metathesis.[10]

Trisamidomolybdenum(VI) alkylidyne complexes catalyze alkyne metathesis.[30]
In the Kauffmann olefination, molybdenum(III) chloride and methyllithium form an organometallic complex capable of carbonyl olefination.[31]
References edit
- ^ Beatrice Roessler; Sven Kleinhenza; Konrad Seppelt (2000). "Pentamethylmolybdenum". Chemical Communications (12): 1039–1040. doi:10.1039/b000987n.
- ^ Poli, R. (2008). "High oxidation state organomolybdenum and organotungsten chemistry in protic environments" (PDF). Coord. Chem. Rev. 252 (15–17): 1592–1612. doi:10.1016/j.ccr.2007.11.029.
- ^ Green M. L. H., Ng D. K. P. (1995). "Cycloheptatriene and -enyl Complexes of the Early Transition Metals". Chemical Reviews. 95 (2): 439–73. doi:10.1021/cr00034a006.
- ^ Ellis, J. E. (2003). "Metal Carbonyl Anions: from [Fe(CO)4]2− to [Hf(CO)6]2− and Beyond". Organometallics. 22 (17): 3322–3338. doi:10.1021/om030105l.
- ^ a b Flower, K. R. (2007). "Molybdenum Compounds without CO or Isonitrile Ligands". In Mingos, D. Michael P.; Crabtree, Robert H. (eds.). Comprehensive Organometallic Chemistry III. Vol. 5. pp. 513–595. doi:10.1016/B0-08-045047-4/00072-8. ISBN 9780080450476.
- ^ Stephan, G. C.; Naether, C.; Peters, G.; Tuczek, F. (2013). "Molybdenum 17- and 18-Electron Bis- and Tris(Butadiene) Complexes: Electronic Structures, Spectroscopic Properties, and Oxidative Ligand Substitution Reactions". Inorg. Chem. 52 (10): 5931–5942. doi:10.1021/ic400145f. PMID 23627292.
- ^ Joseph L. Templeton "Four-Electron Alkyne Ligands in Molybdenum(II) and Tungsten(II) Complexes" Advances in Organometallic Chemistry 1989, Volume 29, Pages 1–100.doi:10.1016/S0065-3055(08)60352-4
- ^ Synthesis of Organometallic Compounds: A Practical Guide Sanshiro Komiya Ed. S. Komiya, M. Hurano 1997
- ^ Kuehn, F. E.; Santos, A. M.; Herrmann, W. A. (2005). "Organorhenium(VII) and Organomolybdenum(VI) Oxides: Syntheses and Application in Olefin Epoxidation". Dalton Trans. (15): 2483–2491. doi:10.1039/b504523a. PMID 16025165.
- ^ a b R.R. Schrock (1986). "High-oxidation-state molybdenum and tungsten alkylidene complexes". Acc. Chem. Res. 19 (11): 342–348. doi:10.1021/ar00131a003.
- ^ McCullough, Laughlin G. (1985). "Multiple metal-carbon bonds. 38. Preparation of trialkoxymolybdenum(VI) alkylidyne complexes, their reactions with acetylenes, and the x-ray structure of Mo[C3(CMe3)2][OCH(CF3)2](C5H5N)2". J. Am. Chem. Soc. 107 (21): 5987. doi:10.1021/ja00307a025.
- ^ Tsai, Yi-Chou; Cummins, Christopher C. (2000). "Facile Synthesis of Trialkoxymolybdenum(VI) Alkylidyne Complexes for Alkyne Metathesis". Organometallics. 19 (25): 5260. doi:10.1021/om000644f.
- ^ Cummins, Christopher C. (1998). "Reductive cleavage and related reactions leading to molybdenum–element multiple bonds: new pathways offered by three-coordinate molybdenum(III)". Chemical Communications (17): 1777–1786. doi:10.1039/A802402B.
- ^ Fürstner, Alois (1999). "Mo[N(t-Bu)(Ar)]3 Complexes As Catalyst Precursors: In Situ Activation and Application to Metathesis Reactions of Alkynes and Diynes". J. Am. Chem. Soc. 121 (40): 9453. doi:10.1021/ja991340r.
- ^ Agapie, Theodor (2002). "Methine (CH) Transfer via a Chlorine Atom Abstraction/Benzene-Elimination Strategy: Molybdenum Methylidyne Synthesis and Elaboration to a Phosphaisocyanide Complex". J. Am. Chem. Soc. 124 (11): 2412–2413. doi:10.1021/ja017278r. PMID 11890770.
- ^ Zhang, Wei; Moore, Jeffrey (2004). "Highly Active Trialkoxymolybdenum(VI) Alkylidyne Catalysts Synthesized by a Reductive Recycle Strategy". J. Am. Chem. Soc. 126 (1): 329–335. doi:10.1021/ja0379868. PMID 14709099.
- ^ Zhang, Wei; Moore, Jeffrey (2004). "Synthesis of Poly(2,5-thienyleneethynylene)s by Alkyne Metathesis". Macromolecules. 37 (11): 3973. Bibcode:2004MaMol..37.3973Z. doi:10.1021/ma049371g.
- ^ Zhang, Wei (2011). "Introducing A Podand Motif to Alkyne Metathesis Catalyst Design: A Highly Active Multidentate Molybdenum(VI) Catalyst that Resists Alkyne Polymerization". Angew. Chem. Int. Ed. 50 (15): 3435–3438. doi:10.1002/anie.201007559. PMID 21394862.
- ^ Fürstner, Alois (2009). "Molybdenum Nitride Complexes with Ph3SiO Ligands Are Exceedingly Practical and Tolerant Precatalysts for Alkyne Metathesis and Efficient Nitrogen Transfer Agents". J. Am. Chem. Soc. 131 (27): 9468–9470. doi:10.1021/ja903259g. PMID 19534524.
- ^ Fürstner, Alois (2010). "Practical New Silyloxy-Based Alkyne Metathesis Catalysts with Optimized Activity and Selectivity Profiles". J. Am. Chem. Soc. 132 (32): 11045–11057. doi:10.1021/ja104800w. PMID 20698671.
- ^ Fürstner, Alois (2013). "Alkyne Metathesis on the Rise". Angew. Chem. Int. Ed. 52 (10): 2794–3519. doi:10.1002/anie.201204513. PMID 23355479.
- ^ Schrock, R. (1978). "Multiple metal-carbon bonds. 12. Tungsten and molybdenum neopentylidyne and some tungsten neopentylidene complexes". J. Am. Chem. Soc. 100 (21): 6774. doi:10.1021/ja00489a049.
- ^ Chisholm, Malcolm H. (2007). "Hexakis(Dimethylamido)Ditungsten and Tungsten(IV) Chloride". Inorganic Syntheses. Vol. 29. pp. 137–140. doi:10.1002/9780470132609.ch33. ISBN 9780470132609.
- ^ Schrock, R. (1982). "Metathesis of tungsten-tungsten triple bonds with acetylenes and nitriles to give alkylidyne and nitrido complexes". J. Am. Chem. Soc. 104 (15): 4291. doi:10.1021/ja00379a061.
- ^ Beer, Stephan (2009). "Experimental and Theoretical Investigations of Catalytic Alkyne Cross-Metathesis with Imidazolin-2-iminato Tungsten Alkylidyne Complexes". Organometallics. 28 (5): 1534. doi:10.1021/om801119t.
- ^ Beer, Stephan (2007). "Efficient Room-Temperature Alkyne Metathesis with Well-Defined Imidazolin-2-iminato Tungsten Alkylidyne Complexes". Angew. Chem. Int. Ed. 46 (46): 8890–8894. doi:10.1002/anie.200703184. PMID 17935104.
- ^ Haberlag, Birte (2010). "Preparation of Imidazolin-2-iminato Molybdenum and Tungsten Benzylidyne Complexes: A New Pathway to Highly Active Alkyne Metathesis Catalysts". Chem. Eur. J. 16 (29): 8868–8877. doi:10.1002/chem.201000597. PMID 20572182.
- ^ Wu, Xian; Daniliuc, Constantin G; Hrib, Cristian G; Tamm, Matthias (2011). "Phosphoraneiminato tungsten alkylidyne complexes as highly efficient alkyne metathesis catalysts". Journal of Organometallic Chemistry. 696 (25): 4147–4151. doi:10.1016/j.jorganchem.2011.06.047. ISSN 0022-328X. OCLC 4925450605.
- ^ Schrock, R. (2007). "Facile Synthesis of a Tungsten Alkylidyne Catalyst for Alkyne Metathesis". Organometallics. 26 (3): 475. doi:10.1021/om0610647.
- ^ Wei Zhang; Yunyi Lu; Jeffrey S. Moore (2007). "Preparation of a Trisamidomolybdenum(VI) Propylidyne Complex". Org. Synth. 84: 163. doi:10.15227/orgsyn.084.0163.Wei Zhang; Hyeon Mo Cho; Jeffrey S. Moore (2007). "Preparation of a Carbazole-Based Macrocycle via Precipitation-driven Alkyne Metathesis" (PDF). Org. Synth. 84: 177. doi:10.15227/orgsyn.084.0177. S2CID 93992722. Archived from the original (PDF) on 2020-02-19.
- ^ Kauffmann, T. (1997). "Organomolybdenum and organotungsten reagents. 7. Novel reactions of organomolybdenum and organotungsten compounds: additive-reductive carbonyl dimerization, spontaneous transformation of methyl ligands into μ-methylene ligands, and selective carbonylmethylenation". Angew. Chem. Int. Ed. Engl. 36: 1259–1275. doi:10.1002/anie.199712581.