


Summary
The P-type ATPases, also known as E1-E2 ATPases, are a large group of evolutionarily related ion and lipid pumps that are found in bacteria, archaea, and eukaryotes.[1] P-type ATPases are α-helical bundle primary transporters named based upon their ability to catalyze auto- (or self-) phosphorylation (hence P) of a key conserved aspartate residue within the pump and their energy source, adenosine triphosphate (ATP). In addition, they all appear to interconvert between at least two different conformations, denoted by E1 and E2.[2] P-type ATPases fall under the P-type ATPase (P-ATPase) Superfamily (TC# 3.A.3) which, as of early 2016, includes 20 different protein families.
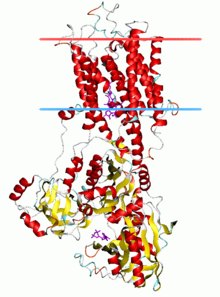 Calcium ATPase, E2-Pi state | |||||||||
| Identifiers | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Symbol | E1-E2_ATPase | ||||||||
| Pfam | PF00122 | ||||||||
| InterPro | IPR008250 | ||||||||
| PROSITE | PDOC00139 | ||||||||
| SCOP2 | 1su4 / SCOPe / SUPFAM | ||||||||
| TCDB | 3.A.3 | ||||||||
| OPM superfamily | 22 | ||||||||
| OPM protein | 3b9b | ||||||||
| Membranome | 224 | ||||||||
| |||||||||
Most members of this transporter superfamily catalyze cation uptake and/or efflux, however one subfamily, the flippases, (TC# 3.A.3.8) is involved in flipping phospholipids to maintain the asymmetric nature of the biomembrane.
In humans, P-type ATPases serve as a basis for nerve impulses, relaxation of muscles, secretion and absorption in the kidney, absorption of nutrient in the intestine and other physiological processes. Prominent examples of P-type ATPases are the sodium-potassium pump (Na+/K+-ATPase), the proton-potassium pump (H+/K+-ATPase), the calcium pump (Ca2+-ATPase) and the plasma membrane proton pump (H+-ATPase) of plants and fungi.
General transport reaction edit
The generalized reaction for P-type ATPases is
nLigand1 (out) + mLigand2 (in) + ATP → nLigand1 (in) + mLigand2 (out) + ADP + Pi.
where the ligand can be either a metal ion or a phospholipid molecule.
Discovery edit
The first P-type ATPase discovered was the Na+/K+-ATPase, which Nobel laureate Jens Christian Skou isolated in 1957.[3] The Na+/K+-ATPase was only the first member of a large and still-growing protein family (see Swiss-Prot Prosite motif PS00154).
Structure edit
P-type ATPases have a single catalytic subunit of 70 - 140 kDa. The catalytic subunit hydrolyzes ATP, contains the aspartyl phosphorylation site and binding sites for the transported ligand(s) and catalyzes ion transport. Various subfamilies of P-type ATPases also need additional subunits for proper function. Additional subunits that lack catalytic activity are present in the ATPase complexes of P1A, P2A, P2C and P4 ATPases. E.g. the catalytic alpha subunit of Na+/K+-ATPase consists of two additional subunits, beta and gamma, involved in trafficking, folding, and regulation of these pumps. The first P-type ATPase to be crystallized was SERCA1a, a sarco(endo)plasmic reticulum Ca2+-ATPase of fast twitch muscle from adult rabbit.[4] It is generally acknowledged that the structure of SERCA1a is representative for the superfamily of P-type ATPases.[5]
The catalytic subunit of P-type ATPases is composed of a cytoplasmic section and a transmembrane section with binding sites for the transported ligand(s). The cytoplasmic section consists of three cytoplasmic domains, designated the P, N, and A domains, containing over half the mass of the protein.
Membrane section edit
The transmembrane section (M domain) typically has ten transmembrane helices (M1-M10), with the binding sites for transported ligand(s) located near the midpoint of the bilayer. While most subfamilies have 10 transmembrane helices, there are some notable exceptions. The P1A ATPases are predicted to have 7, and the large subfamily of heavy metal pumps P1B) is predicted to have 8 transmembrane helices. P5 ATPases appear to have a total of 12 transmembrane helices.
Common for all P-type ATPases is a core of 6 transmembrane-spanning segments (also called the 'transport (T) domain'; M1-M6 in SERCA), that harbors the binding sites for the translocated ligand(s). The ligand(s) enter through a half-channel to the binding site and leave on the other side of the membrane through another half-channel.
Varying among P-type ATPase is the additional number of transmembrane-spanning segments (also called the 'support (S) domain', which between subfamilies ranges from 2 to 6. Extra transmembrane-segments likely provides structural support for the T domain and can also have specialized functions.
Phosphorylation (P) domain edit
The P domain contains the canonical aspartic acid residue phosphorylated (in a conserved DKTGT motif; the 'D' is the one letter abbreviation of the amino acid aspartate) during the reaction cycle. It is composed of two parts widely separated in sequence. These two parts assemble into a seven-strand parallel β-sheet with eight short associated a-helices, forming a Rossmann fold.
The folding pattern and the locations of the critical amino acids for phosphorylation in P-type ATPases has the haloacid dehalogenase fold characteristic of the haloacid dehalogenase (HAD) superfamily, as predicted by sequence homology. The HAD superfamily functions on the common theme of an aspartate ester formation by an SN2 reaction mechanism. This SN2 reaction is clearly observed in the solved structure of SERCA with ADP plus AlF4−.[6]
Nucleotide binding (N) domain edit
The N domain serves as a built-in protein kinase that functions to phosphorylate the P domain. The N domain is inserted between the two segments of the P domain, and is formed of a seven-strand antiparallel β-sheet between two helix bundles. This domain contains the ATP-binding pocket, pointing out toward the solvent near the P-domain.
Actuator (A) domain edit
The A domain serves as a built-in protein phosphatase that functions to dephosphorylate the phosphorylated P domain. The A domain is the smallest of the three cytoplasmic domains. It consists of a distorted jellyroll structure and two short helices. It is the actuator domain modulating the occlusion of the transported ligand(s) in the transmembrane binding sites, and it is pivot in transposing the energy from the hydrolysis of ATP in the cytoplasmic domains to the vectorial transport of cations in the transmembrane domain. The A domain dephosphorylates the P domain as part of the reaction cycle using a highly conserved TGES motif located at one end of the jellyroll.
Regulatory (R) domain edit
Some members of the P-type ATPase family have additional regulatory (R) domains fused to the pump. Heavy metal P1B pumps can have several N- and C-terminal heavy metal-binding domains that have been found to be involved in regulation. The P2B Ca2+ ATPases have autoinbitory domains in their amino-terminal (plants) or carboxy-terminal (animals) regions, which contain binding sites for calmodulin, which, in the presence of Ca2+, activates P2B ATPases by neutralizing the terminal constraint. The P3A plasma membrane proton pumps have a C-terminal regulatory domain, which, when unphosphorylated, inhibits pumping.
Mechanism edit
All P-type ATPases use the energy derived from ATP to drive transport. They form a high-energy aspartyl-phosphoanhydride intermediate in the reaction cycle, and they interconvert between at least two different conformations, denoted by E1 and E2. The E1-E2 notation stems from the initial studies on this family of enzymes made on the Na+/K+-ATPase, where the sodium form and the potassium form are referred to as E1 and E2, respectively, in the "Post-Albers scheme". The E1-E2 schema has been proven to work, but there exist more than two major conformational states. The E1-E2 notation highlights the selectivity of the enzyme. In E1, the pump has high affinity for the exported substrate and low affinity for the imported substrate. In E2, it has low affinity of the exported substrate and high affinity for the imported substrate. Four major enzyme states form the cornerstones in the reaction cycle. Several additional reaction intermediates occur interposed. These are termed E1~P, E2P, E2-P*, and E1/E2.[7]
ATP hydrolysis occurs in the cytoplasmic headpiece at the interface between domain N and P. Two Mg-ion sites form part of the active site. ATP hydrolysis is tightly coupled to translocation of the transported ligand(s) through the membrane, more than 40 Å away, by the A domain.
Classification edit
A phylogenetic analysis of 159 sequences made in 1998 by Axelsen and Palmgren suggested that P-type ATPases can be divided into five subfamilies (types; designated as P1-P5), based strictly on a conserved sequence kernel excluding the highly variable N and C terminal regions.[8] Chan et al. (2010) also analyzed P-type ATPases in all major prokaryotic phyla for which complete genome sequence data were available and compared the results with those for eukaryotic P-type ATPases.[9] The phylogenetic analysis grouped the proteins independent of the organism from which they are isolated and showed that the diversification of the P-type ATPase family occurred prior to the separation of eubacteria, archaea, and eucaryota. This underlines the significance of this protein family for cell survival under stress conditions.[8]
P1 ATPases edit
P1 ATPases (or Type I ATPases) consists of the transition/heavy metal ATPases. Topological type I (heavy metal) P-type ATPases predominate in prokaryotes (approx. tenfold).[10]
P1A ATPases (potassium pumps) edit
P1A ATPases (or Type IA) are involved in K+ import (TC# 3.A.3.7). They are atypical P-type ATPases because, unlike other P-type ATPases, they function as part of a heterotetrameric complex (called KdpFABC), where the actual K+ transport is mediated by another subcomponent of the complex.
P1B ATPases (heavy metal pumps) edit
P1B ATPases (or Type IB ATPases) are involved in transport of the soft Lewis acids: Cu+, Ag+, Cu2+, Zn2+, Cd2+, Pb2+ and Co2+ (TC#s 3.A.3.5 and 3.A.3.6). They are key elements for metal resistance and metal homeostasis in a wide range of organisms.
Metal binding to transmembrane metal-binding sites (TM-MBS) in Cu+-ATPases is required for enzyme phosphorylation and subsequent transport. However, Cu+ does not access Cu+-ATPases in a free (hydrated) form but is bound to a chaperone protein. The delivery of Cu+ by Archaeoglobus fulgidus Cu+-chaperone, CopZ (see TC# 3.A.3.5.7), to the corresponding Cu+-ATPase, CopA (TC# 3.A.3.5.30), has been studied.[11] CopZ interacted with and delivered the metal to the N-terminal metal binding domain(s) of CopA (MBDs). Cu+-loaded MBDs, acting as metal donors, were unable to activate CopA or a truncated CopA lacking MBDs. Conversely, Cu+-loaded CopZ activated the CopA ATPase and CopA constructs in which MBDs were rendered unable to bind Cu+. Furthermore, under nonturnover conditions, CopZ transferred Cu+ to the TM-MBS of a CopA lacking MBDs altogether. Thus, MBDs may serve a regulatory function without participating directly in metal transport, and the chaperone delivers Cu+ directly to transmembrane transport sites of Cu+-ATPases.[11] Wu et al. (2008) have determined structures of two constructs of the Cu (CopA) pump from Archaeoglobus fulgidus by cryoelectron microscopy of tubular crystals, which revealed the overall architecture and domain organization of the molecule. They localized its N-terminal MBD within the cytoplasmic domains that use ATP hydrolysis to drive the transport cycle and built a pseudoatomic model by fitting existing crystallographic structures into the cryoelectron microscopy maps for CopA. The results also similarly suggested a Cu-dependent regulatory role for the MBD.[12]
In the Archaeoglobus fulgidus CopA (TC# 3.A.3.5.7), invariant residues in helixes 6, 7 and 8 form two transmembrane metal binding sites (TM-MBSs). These bind Cu+ with high affinity in a trigonal planar geometry. The cytoplasmic Cu+ chaperone CopZ transfers the metal directly to the TM-MBSs; however, loading both of the TM-MBSs requires binding of nucleotides to the enzyme. In agreement with the classical transport mechanism of P-type ATPases, occupancy of both transmembrane sites by cytoplasmic Cu+ is a requirement for enzyme phosphorylation and subsequent transport into the periplasmic or extracellular milieu. Transport studies have shown that most Cu+-ATPases drive cytoplasmic Cu+ efflux, albeit with quite different transport rates in tune with their various physiological roles. Archetypical Cu+-efflux pumps responsible for Cu+ tolerance, like the Escherichia coli CopA, have turnover rates ten times higher than those involved in cuproprotein assembly (or alternative functions). This explains the incapability of the latter group to significantly contribute to the metal efflux required for survival in high copper environments. Structural and mechanistic details of copper-transporting P-type ATPase functionhave been described.[13]
P2 ATPases edit
P2 ATPases (or Type II ATPases) are split into four groups. Topological type II ATPases (specific for Na+,K+, H+ Ca2+, Mg2+ and phospholipids) predominate in eukaryotes (approx. twofold).[10]
P2A ATPases (calcium pumps) edit
P2A ATPases (or Type IIA ATPases) are Ca2+ ATPases that transport Ca2+. P2A ATPases are split into two groups. Members of the first group are called sarco/endoplasmatic reticulum Ca2+-ATPases (also referred to as SERCA). These pumps have two Ca2+ ion binding sites and are often regulated by inhibitory accessory proteins having a single trans-membrane spanning segment (e.g.phospholamban and sarcolipin. In the cell, they are located in the sarcoplasmic or endoplasmatic reticulum. SERCA1a is a type IIA pump. The second group of P2A ATPases is called secretory pathway Ca2+-ATPases (also referred to as SPCA). These pumps have a single Ca2+ ion binding site and are located in secretory vesicles (animals) or the vacuolar membrane (fungi). (TC# 3.A.3.2)
Crystal structures of Sarcoplasimc/endoplasmic reticulum ATP driven calcium pumps can be found in RCSB.[14]
SERCA1a is composed of a cytoplasmic section and a transmembrane section with two Ca2+-binding sites. The cytoplasmic section consists of three cytoplasmic domains, designated the P, N, and A domains, containing over half the mass of the protein. The transmembrane section has ten transmembrane helices (M1-M10), with the two Ca2+-binding sites located near the midpoint of the bilayer. The binding sites are formed by side-chains and backbone carbonyls from M4, M5, M6, and M8. M4 is unwound in this region due to a conserved proline (P308). This unwinding of M4 is recognised as a key structural feature of P-type ATPases.
Structures are available for both the E1 and E2 states of the Ca2+ ATPase showing that Ca2+ binding induces major changes in all three cytoplasmic domains relative to each other.[15]
In the case of SERCA1a, energy from ATP is used to transport 2 Ca2+-ions from the cytoplasmic side to the lumen of the sarcoplasmatic reticulum, and to countertransport 1-3 protons into the cytoplasm. Starting in the E1/E2 state, the reaction cycle begins as the enzyme releases 1-3 protons from the cation-ligating residues, in exchange for cytoplasmic Ca2+-ions. This leads to assembly of the phosphorylation site between the ATP-bound N domain and the P domain, while the A domain directs the occlusion of the bound Ca2+. In this occluded state, the Ca2+ ions are buried in a proteinaceous environment with no access to either side of the membrane. The Ca2E1~P state becomes formed through a kinase reaction, where the P domain becomes phosphorylated, producing ADP. The cleavage of the β-phosphodiester bond releases the gamma-phosphate from ADP and unleashes the N domain from the P domain.
This then allows the A domain to rotate toward the phosphorylation site, making a firm association with both the P and the N domains. This movement of the A domain exerts a downward push on M3-M4 and a drag on M1-M2, forcing the pump to open at the luminal side and forming the E2P state. During this transition, the transmembrane Ca2+-binding residues are forced apart, destroying the high-affinity binding site. This is in agreement with the general model form substrate translocation, showing that energy in primary transport is not used to bind the substrate but to release it again from the buried counter ions. At the same time the N domain becomes exposed to the cytosol, ready for ATP exchange at the nucleotide-binding site.
As the Ca2+ dissociate to the luminal side, the cation binding sites are neutralised by proton binding, which makes a closure of the transmembrane segments favourable. This closure is coupled to a downward rotation of the A domain and a movement of the P domain, which then leads to the E2-P* occluded state. Meanwhile, the N domain exchanges ADP for ATP.
The P domain is dephosphorylated by the A domain, and the cycle completes when the phosphate is released from the enzyme, stimulated by the newly bound ATP, while a cytoplasmic pathway opens to exchange the protons for two new Ca2+ ions.[7]
Xu et al. proposed how Ca2+ binding induces conformational changes in TMS 4 and 5 in the membrane domain (M) that in turn induce rotation of the phosphorylation domain (P).[15] The nucleotide binding (N) and β-sheet (β) domains are highly mobile, with N flexibly linked to P, and β flexibly linked to M. Modeling of the fungal H+ ATPase, based on the structures of the Ca2+ pump, suggested a comparable 70º rotation of N relative to P to deliver ATP to the phosphorylation site.[16]
One report suggests that this sarcoplasmic reticulum (SR) Ca2+ ATPase is homodimeric.[17]
Crystal structures have shown that the conserved TGES loop of the Ca2+-ATPase is isolated in the Ca2E1 state but becomes inserted in the catalytic site in E2 states.[18] Anthonisen et al. (2006) characterized the kinetics of the partial reaction steps of the transport cycle and the binding of the phosphoryl analogs BeF, AlF, MgF, and vanadate in mutants with alterations to conserved TGES loop residues. The data provide functional evidence supporting a role of Glu183 in activating the water molecule involved in the E2P → E2 dephosphorylation and suggest a direct participation of the side chains of the TGES loop in the control and facilitation of the insertion of the loop in the catalytic site. The interactions of the TGES loop furthermore seem to facilitate its disengagement from the catalytic site during the E2 → Ca2E1 transition.[18]
Crystal Structures of Calcium ATPase are available in RCSB and include: PDB: 4AQR, 2L1W, 2M7E, 2M73, among others.[19]
P2B ATPases (calcium pumps) edit
P2B (or Type IIB ATPases) are Ca2+ ATPases that transport Ca2+. These pumps have a single Ca2+ ion binding site and are regulated by binding of calmodulin to autoinhibitory built-in domains situated at either the carboxy-terminal (animals) or amino-terminal (plants) end of the pump protein. In the cell, they are situated in the plasma membrane (animals and plants) and the internal membranes (plants). Plasma membrane Ca2+-ATPase (PMCA) of animals is a P2B ATPase (TC# 3.A.3.2)
P2C ATPases (sodium/potassium and proton/potassium pumps) edit
P2C ATPases (or Type IIC) include the closely related Na+/K+ and H+/K+ ATPases from animal cells. (TC# 3.A.3.1)
The X-ray crystal structure at 3.5 Å resolution of the pig renal Na+/K+-ATPase has been determined with two rubidium ions bound in an occluded state in the transmembrane part of the α-subunit.[20] Several of the residues forming the cavity for rubidium/potassium occlusion in the Na+/K+-ATPase are homologous to those binding calcium in the Ca2+-ATPase of the sarco(endo)plasmic reticulum. The carboxy terminus of the α-subunit is contained within a pocket between transmembrane helices and seems to be a novel regulatory element controlling sodium affinity, possibly influenced by the membrane potential.
Crystal Structures are available in RCSB and include: PDB: 4RES, 4RET, 3WGU, 3WGV, among others.[21]
P2D ATPases (sodium pumps) edit
P2D ATPases (or Type IID) include a small number of Na+ (and K+) exporting ATPases found in fungi and mosses. (Fungal K+ transporters; TC# 3.A.3.9)
P3 ATPases edit
P3 ATPases (or Type III ATPases) are split into two groups.
P3A ATPases (proton pumps) edit
P3A ATPases (or Type IIIA) contain the plasma membrane H+-ATPases from prokaryotes, protists, plants and fungi.
Plasma membrane H+-ATPase is best characterized in plants and yeast. It maintains the level of intracellular pH and transmembrane potential.[22] Ten transmembrane helices and three cytoplasmic domains define the functional unit of ATP-coupled proton transport across the plasma membrane, and the structure is locked in a functional state not previously observed in P-type ATPases. The transmembrane domain reveals a large cavity, which is likely to be filled with water, located near the middle of the membrane plane where it is lined by conserved hydrophilic and charged residues. Proton transport against a high membrane potential is readily explained by this structural arrangement.[23]
P3B ATPases (magnesium pumps) edit
P3B ATPases (or Type IIIB) are presumed Mg2+-ATPases found in eubacteria and plants. Fungal H+ transporters (TC# 3.A.3.3) and Mg2+ (TC# 3.A.3.4)
P4 ATPases (phospholipid flippases) edit
P4 ATPases (or Type IV ATPases) are flippases involved in the transport of phospholipids,[24] such as phosphatidylserine, phosphatidylcholine and phosphatidylethanolamine.[25]
P5 ATPases edit
P5 ATPases (or Type V ATPases) have unknown specificity. This large group is found only in eukaryotes and is further divided into two groups.
P5A ATPases edit
P5A ATPases (or Type VA) are involved in regulation of homeostasis in the endoplasmic reticulum.[26]
P5B ATPases edit
P5B ATPases (or Type VB) are found in the lysosomal membrane of animals. Mutations in these pumps are linked to a variety of neurological diseases.[27][28]
Further phylogenetic classification edit
In addition to the subfamilies of P-type ATPases listed above, several prokaryotic families of unknown function have been identified.[29] The Transporter Classification Database provides a representative list of members of the P-ATPase superfamily, which as of early 2016 consisting of 20 families. Members of the P-ATPase superfamily are found in bacteria, archaea and eukaryotes. Clustering on the phylogenetic tree is usually in accordance with specificity for the transported ion(s).
In eukaryotes, they are present in the plasma membranes or endoplasmic reticular membranes. In prokaryotes, they are localized to the cytoplasmic membranes.
P-type ATPases from 26 eukaryotic species were analyzed later.[10][30]
Chan et al., (2010) conducted an equivalent but more extensive analysis of the P-type ATPase Superfamily in Prokaryotes and compared them with those from Eukaryotes. While some families are represented in both types of organisms, others are found only in one of the other type. The primary functions of prokaryotic P-type ATPases appear to be protection from environmental stress conditions. Only about half of the P-type ATPase families are functionally characterized.[29]
Horizontal Gene Transfer edit
Many P-type ATPase families are found exclusively in prokaryotes (e.g. Kdp-type K+ uptake ATPases (type III) and all prokaryotic functionally uncharacterized P-type ATPase (FUPA) families), while others are restricted to eukaryotes (e.g. phospholipid flippases and all 13 eukaryotic FUPA families).[10] Horizontal gene transfer has occurred frequently among bacteria and archaea, which have similar distributions of these enzymes, but rarely between most eukaryotic kingdoms, and even more rarely between eukaryotes and prokaryotes. In some bacterial phyla (e.g. Bacteroidota and Fusobacteriota), ATPase gene gain and loss as well as horizontal transfer occurred seldom in contrast to most other bacterial phyla. Some families (i.e., Kdp-type ATPases) underwent far less horizontal gene transfer than other prokaryotic families, possibly due to their multisubunit characteristics. Functional motifs are better conserved across family lines than across organismal lines, and these motifs can be family specific, facilitating functional predictions. In some cases, gene fusion events created P-type ATPases covalently linked to regulatory catalytic enzymes. In one family (FUPA Family 24), a type I ATPase gene (N-terminal) is fused to a type II ATPase gene (C-terminal) with retention of function only for the latter. Genome minimalization led to preferential loss of P-type ATPase genes. Chan et al. (2010) suggested that in prokaryotes and some unicellular eukaryotes, the primary function of P-type ATPases is protection from extreme environmental stress conditions. The classification of P-type ATPases of unknown function into phylogenetic families provides guides for future molecular biological studies.[9]
Human genes edit
Human genes encoding P-type ATPases or P-type ATPase-like proteins include:
- P1B: Cu++ ATPase: ATP7A, ATP7B
- P2A: SERCA Ca2+ ATPase: ATP2A1, ATP2A2, ATP2A3
- P2A: secretory pathway Ca2+-ATPase: ATP2C1, ATP2C2
- P2B: Ca2+ ATPase: ATP2B1, ATP2B2, ATP2B3, ATP2B4
- P2C: Na+/K+ ATPase: ATP1A1, ATP1A2, ATP1A3, ATP1A4, ATP1B1, ATP1B2, ATP1B3, ATP1B4
- P2C: H+/K+ ATPase, gastric: ATP4A;
- P2C: H+/K+ ATPase, nongastric: ATP12A
- P4: Flippase: ATP8A1, ATP8B1, ATP8B2, ATP8B3, ATP8B4, ATP9A, ATP9B, ATP10A, ATP10B, ATP10D, ATP11A, ATP11B, ATP11C
- P5: ATP13A1, ATP13A2, ATP13A3, ATP13A4, ATP13A5
See also edit
References edit
- ^ Palmgren MG, Nissen P (2011). "P-type ATPases" (PDF). Annu. Rev. Biophys. 40: 243–66. doi:10.1146/annurev.biophys.093008.131331. PMID 21351879.
- ^ Pedersen PL, Carafoli E (1987). "Ion motive ATPases. I. Ubiquity, properties, and significance to cell function". Trends in Biochemical Sciences. 12: 146–50. doi:10.1016/0968-0004(87)90071-5.
- ^ SKOU JC (February 1957). "The influence of some cations on an adenosine triphosphatase from peripheral nerves". Biochim. Biophys. Acta. 23 (2): 394–401. doi:10.1016/0006-3002(57)90343-8. PMID 13412736.
- ^ Toyoshima C, Nakasako M, Nomura H, Ogawa H (June 2000). "Crystal structure of the calcium pump of sarcoplasmic reticulum at 2.6 A resolution". Nature. 405 (6787): 647–55. Bibcode:2000Natur.405..647T. doi:10.1038/35015017. PMID 10864315. S2CID 4316039.
- ^ Stokes DL, Green NM (2003). "Structure and function of the calcium pump". Annu Rev Biophys Biomol Struct. 32: 445–68. doi:10.1146/annurev.biophys.32.110601.142433. PMID 12598367.
- ^ PDB: 1T5T; Sørensen TL, Møller JV, Nissen P (June 2004). "Phosphoryl transfer and calcium ion occlusion in the calcium pump". Science. 304 (5677): 1672–5. Bibcode:2004Sci...304.1672S. doi:10.1126/science.1099366. PMID 15192230. S2CID 30576015.
- ^ a b Olesen C, Picard M, Winther AM, et al. (December 2007). "The structural basis of calcium transport by the calcium pump". Nature. 450 (7172): 1036–42. Bibcode:2007Natur.450.1036O. doi:10.1038/nature06418. PMID 18075584. S2CID 4323780.
- ^ a b Axelsen KB, Palmgren MG (January 1998). "Evolution of substrate specificities in the P-type ATPase superfamily". J. Mol. Evol. 46 (1): 84–101. Bibcode:1998JMolE..46...84A. doi:10.1007/PL00006286. PMID 9419228. S2CID 10238525. Archived from the original on 2000-09-15. Retrieved 2009-06-10.
- ^ a b Chan, Henry; Babayan, Vartan; Blyumin, Elya; Gandhi, Charmy; Hak, Kunal; Harake, Danielle; Kumar, Kris; Lee, Perry; Li, Tze T. (2010). "The P-Type ATPase Superfamily". Journal of Molecular Microbiology and Biotechnology. 19 (1–2): 5–104. doi:10.1159/000319588. PMID 20962537. S2CID 7316282.
- ^ a b c d Thever, Mark D.; Jr, Milton H. Saier (2009-06-23). "Bioinformatic Characterization of P-Type ATPases Encoded Within the Fully Sequenced Genomes of 26 Eukaryotes". Journal of Membrane Biology. 229 (3): 115–130. doi:10.1007/s00232-009-9176-2. ISSN 0022-2631. PMC 2709905. PMID 19548020.
- ^ a b González-Guerrero, Manuel; Argüello, José M. (2008-04-22). "Mechanism of Cu+-transporting ATPases: soluble Cu+ chaperones directly transfer Cu+ to transmembrane transport sites". Proceedings of the National Academy of Sciences of the United States of America. 105 (16): 5992–5997. Bibcode:2008PNAS..105.5992G. doi:10.1073/pnas.0711446105. ISSN 1091-6490. PMC 2329688. PMID 18417453.
- ^ Wu, Chen-Chou; Rice, William J.; Stokes, David L. (2008-06-01). "Structure of a copper pump suggests a regulatory role for its metal-binding domain". Structure. 16 (6): 976–985. doi:10.1016/j.str.2008.02.025. ISSN 0969-2126. PMC 2705936. PMID 18547529.
- ^ Meng, Dan; Bruschweiler-Li, Lei; Zhang, Fengli; Brüschweiler, Rafael (2015-08-18). "Modulation and Functional Role of the Orientations of the N- and P-Domains of Cu+ -Transporting ATPase along the Ion Transport Cycle". Biochemistry. 54 (32): 5095–5102. doi:10.1021/acs.biochem.5b00420. ISSN 1520-4995. PMID 26196187.
- ^ "Rcsb Pdb".
- ^ a b Xu, Chen; Rice, William J.; He, Wanzhong; Stokes, David L. (2002-02-08). "A structural model for the catalytic cycle of Ca(2+)-ATPase". Journal of Molecular Biology. 316 (1): 201–211. doi:10.1006/jmbi.2001.5330. ISSN 0022-2836. PMID 11829513. S2CID 596014.
- ^ Kühlbrandt, Werner; Zeelen, Johan; Dietrich, Jens (2002-09-06). "Structure, mechanism, and regulation of the Neurospora plasma membrane H+-ATPase". Science. 297 (5587): 1692–1696. Bibcode:2002Sci...297.1692K. doi:10.1126/science.1072574. ISSN 1095-9203. PMID 12169656. S2CID 16320388.
- ^ Ushimaru, Makoto; Fukushima, Yoshihiro (2008-09-15). "The dimeric form of Ca2+-ATPase is involved in Ca2+ transport in the sarcoplasmic reticulum". The Biochemical Journal. 414 (3): 357–361. doi:10.1042/BJ20071701. ISSN 1470-8728. PMID 18471093. S2CID 698714.
- ^ a b Anthonisen, Anne Nyholm; Clausen, Johannes D.; Andersen, Jens Peter (2006-10-20). "Mutational analysis of the conserved TGES loop of sarcoplasmic reticulum Ca2+-ATPase". The Journal of Biological Chemistry. 281 (42): 31572–31582. doi:10.1074/jbc.M605194200. ISSN 0021-9258. PMID 16893884.
- ^ "Rcsb Pdb".
- ^ Morth, J. Preben; Pedersen, Bjørn P.; Toustrup-Jensen, Mads S.; Sørensen, Thomas L.-M.; Petersen, Janne; Andersen, Jens Peter; Vilsen, Bente; Nissen, Poul (2007-12-13). "Crystal structure of the sodium-potassium pump". Nature. 450 (7172): 1043–1049. Bibcode:2007Natur.450.1043M. doi:10.1038/nature06419. ISSN 1476-4687. PMID 18075585. S2CID 4344526.
- ^ "Rcsb Pdb".
- ^ Kühlbrandt, Werner; Zeelen, Johan; Dietrich, Jens (2002-09-06). "Structure, Mechanism, and Regulation of the Neurospora Plasma Membrane H+-ATPase". Science. 297 (5587): 1692–1696. Bibcode:2002Sci...297.1692K. doi:10.1126/science.1072574. ISSN 0036-8075. PMID 12169656. S2CID 16320388.
- ^ Pedersen, Bjørn P.; Buch-Pedersen, Morten J.; Preben Morth, J.; Palmgren, Michael G.; Nissen, Poul (2007-12-13). "Crystal structure of the plasma membrane proton pump". Nature. 450 (7172): 1111–1114. Bibcode:2007Natur.450.1111P. doi:10.1038/nature06417. ISSN 0028-0836. PMID 18075595. S2CID 4413142.
- ^ Lenoir G, Williamson P, Holthuis JC (December 2007). "On the origin of lipid asymmetry: the flip side of ion transport". Curr Opin Chem Biol. 11 (6): 654–61. doi:10.1016/j.cbpa.2007.09.008. hdl:1874/26974. PMID 17981493.
- ^ Lopez-Marques RL, Poulsen LR, Hanisch S, Meffert K, Buch-Pedersen MJ, Jakobsen MK, Pomorski TG, Palmgren MG (2010). "Intracellular targeting signals and lipid specificity determinants of the ALA/ALIS P4-ATPase complex reside in the catalytic ALA alpha-subunit". Mol Biol Cell. 21 (5): 791–801. doi:10.1091/mbc.E09-08-0656. PMC 2828965. PMID 20053675.
- ^ Sørensen DM, Holen HW, Holemans T, Vangheluwe P, Palmgren MG (May 2014). "Towards defining the substrate of orphan P5A-ATPases" (PDF). Biochim. Biophys. Acta. 1850 (3): 524–35. doi:10.1016/j.bbagen.2014.05.008. PMID 24836520.
- ^ Ramirez, A; Heimbach, A; Gründemann, J; Stiller, B; Hampshire, D; Cid, L. P; Goebel, I; Mubaidin, A. F; Wriekat, A. L; Roeper, J; Al-Din, A; Hillmer, A. M; Karsak, M; Liss, B; Woods, C. G; Behrens, M. I; Kubisch, C (2006). "Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase". Nature Genetics. 38 (10): 1184–91. doi:10.1038/ng1884. PMID 16964263. S2CID 6502952.
- ^ Di Fonzo, A; Chien, H. F; Socal, M; Giraudo, S; Tassorelli, C; Iliceto, G; Fabbrini, G; Marconi, R; Fincati, E; Abbruzzese, G; Marini, P; Squitieri, F; Horstink, M. W; Montagna, P; Libera, A. D; Stocchi, F; Goldwurm, S; Ferreira, J. J; Meco, G; Martignoni, E; Lopiano, L; Jardim, L. B; Oostra, B. A; Barbosa, E. R; Italian Parkinson Genetics Network; Bonifati, V (2007). "ATP13A2 missense mutations in juvenile parkinsonism and young onset Parkinson disease". Neurology. 68 (19): 1557–62. doi:10.1212/01.wnl.0000260963.08711.08. PMID 17485642. S2CID 24070567.
- ^ a b Chan, Henry; Babayan, Vartan; Blyumin, Elya; Gandhi, Charmy; Hak, Kunal; Harake, Danielle; Kumar, Kris; Lee, Perry; Li, Tze T. (2010-01-01). "The p-type ATPase superfamily". Journal of Molecular Microbiology and Biotechnology. 19 (1–2): 5–104. doi:10.1159/000319588. ISSN 1660-2412. PMID 20962537. S2CID 7316282.
- ^ Rodríguez-Navarro, Alonso; Benito, Begoña (2010-10-01). "Sodium or potassium efflux ATPase: A fungal, bryophyte, and protozoal ATPase". Biochimica et Biophysica Acta (BBA) - Biomembranes. 1798 (10): 1841–1853. doi:10.1016/j.bbamem.2010.07.009. PMID 20650263.