


Summary
Phenylalanine (symbol Phe or F)[3] is an essential α-amino acid with the formula C
9H
11NO
2. It can be viewed as a benzyl group substituted for the methyl group of alanine, or a phenyl group in place of a terminal hydrogen of alanine. This essential amino acid is classified as neutral, and nonpolar because of the inert and hydrophobic nature of the benzyl side chain. The L-isomer is used to biochemically form proteins coded for by DNA. Phenylalanine is a precursor for tyrosine, the monoamine neurotransmitters dopamine, norepinephrine (noradrenaline), and epinephrine (adrenaline), and the biological pigment melanin. It is encoded by the messenger RNA codons UUU and UUC.
 Skeletal formula of L-phenylalanine
| |||
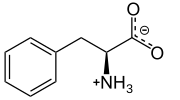 L-Phenylalanine at physiological pH
| |||
| |||
| Names | |||
|---|---|---|---|
| Pronunciation | US: /ˌfɛnəlˈæləniːn/; UK: /ˌfiːnaɪl-/ | ||
| IUPAC name
Phenylalanine
| |||
| Systematic IUPAC name
(S)-2-Amino-3-phenylpropanoic acid | |||
| Identifiers | |||
| |||
3D model (JSmol)
|
| ||
| ChEBI |
| ||
| ChEMBL |
| ||
| ChemSpider |
| ||
| DrugBank |
| ||
| ECHA InfoCard | 100.000.517 | ||
| |||
| KEGG |
| ||
PubChem CID
|
| ||
| UNII |
| ||
CompTox Dashboard (EPA)
|
| ||
| |||
| |||
| Properties | |||
| C9H11NO2 | |||
| Molar mass | 165.192 g·mol−1 | ||
| 9.97 g/L at 0 °C 14.11 g/L at 25 °C | |||
| Acidity (pKa) | 1.83 (carboxyl), 9.13 (amino)[2] | ||
| Hazards | |||
| NFPA 704 (fire diamond) | |||
| Supplementary data page | |||
| Phenylalanine (data page) | |||
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).
Infobox references
| |||


Phenylalanine is found naturally in the milk of mammals. It is used in the manufacture of food and drink products and sold as a nutritional supplement as it is a direct precursor to the neuromodulator phenethylamine. As an essential amino acid, phenylalanine is not synthesized de novo in humans and other animals, who must ingest phenylalanine or phenylalanine-containing proteins.
The one-letter symbol F was assigned to phenylalanine for its phonetic similarity.[4]
History edit
The first description of phenylalanine was made in 1879, when Schulze and Barbieri identified a compound with the empirical formula, C9H11NO2, in yellow lupine (Lupinus luteus) seedlings. In 1882, Erlenmeyer and Lipp first synthesized phenylalanine from phenylacetaldehyde, hydrogen cyanide, and ammonia.[5][6]
The genetic codon for phenylalanine was first discovered by J. Heinrich Matthaei and Marshall W. Nirenberg in 1961. They showed that by using mRNA to insert multiple uracil repeats into the genome of the bacterium E. coli, they could cause the bacterium to produce a polypeptide consisting solely of repeated phenylalanine amino acids. This discovery helped to establish the nature of the coding relationship that links information stored in genomic nucleic acid with protein expression in the living cell.
Dietary sources edit
Good sources of phenylalanine are eggs, chicken, liver, beef, milk, and soybeans.[7] Another common source of phenylalanine is anything sweetened with the artificial sweetener aspartame, such as diet drinks, diet foods and medication; the metabolism of aspartame produces phenylalanine as one of the compound's metabolites.[8]
Dietary recommendations edit
The Food and Nutrition Board (FNB) of the U.S. Institute of Medicine set Recommended Dietary Allowances (RDAs) for essential amino acids in 2002. For phenylalanine plus tyrosine, for adults 19 years and older, 33 mg/kg body weight/day.[9] In 2005 the DRI is set to 27 mg/kg per day (with no tyrosine), the FAO/WHO/UNU recommendation of 2007 is 25 mg/kg per day (with no tyrosine).[10]
Other biological roles edit
L-Phenylalanine is biologically converted into L-tyrosine, another one of the DNA-encoded amino acids. L-tyrosine in turn is converted into L-DOPA, which is further converted into dopamine, norepinephrine (noradrenaline), and epinephrine (adrenaline). The latter three are known as the catecholamines.
Phenylalanine uses the same active transport channel as tryptophan to cross the blood–brain barrier. In excessive quantities, supplementation can interfere with the production of serotonin and other aromatic amino acids[11] as well as nitric oxide due to the overuse (eventually, limited availability) of the associated cofactors, iron or tetrahydrobiopterin. [citation needed] The corresponding enzymes for those compounds are the aromatic amino acid hydroxylase family and nitric oxide synthase.
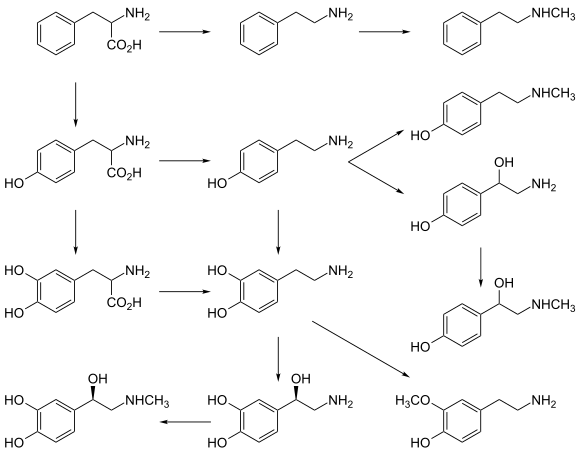
In plants edit
Phenylalanine is the starting compound used in the synthesis of flavonoids. Lignan is derived from phenylalanine and from tyrosine. Phenylalanine is converted to cinnamic acid by the enzyme phenylalanine ammonia-lyase.[15]
Biosynthesis edit
Phenylalanine is biosynthesized via the shikimate pathway.
Phenylketonuria edit
The genetic disorder phenylketonuria (PKU) is the inability to metabolize phenylalanine because of a lack of the enzyme phenylalanine hydroxylase. Individuals with this disorder are known as "phenylketonurics" and must regulate their intake of phenylalanine. Phenylketonurics often use blood tests to monitor the amount of phenylalanine in their blood. Lab results may report phenylalanine levels using either mg/dL and μmol/L. One mg/dL of phenylalanine is approximately equivalent to 60 μmol/L.
A (rare) "variant form" of phenylketonuria called hyperphenylalaninemia is caused by the inability to synthesize a cofactor called tetrahydrobiopterin, which can be supplemented. Pregnant women with hyperphenylalaninemia may show similar symptoms of the disorder (high levels of phenylalanine in blood), but these indicators will usually disappear at the end of gestation. Pregnant women with PKU must control their blood phenylalanine levels even if the fetus is heterozygous for the defective gene because the fetus could be adversely affected due to hepatic immaturity.[medical citation needed]
A non-food source of phenylalanine is the artificial sweetener aspartame. This compound is metabolized by the body into several chemical byproducts including phenylalanine. The breakdown problems phenylketonurics have with the buildup of phenylalanine in the body also occurs with the ingestion of aspartame, although to a lesser degree. Accordingly, all products in Australia, the U.S. and Canada that contain aspartame must be labeled: "Phenylketonurics: Contains phenylalanine." In the UK, foods containing aspartame must carry ingredient panels that refer to the presence of "aspartame or E951"[16] and they must be labeled with a warning "Contains a source of phenylalanine." In Brazil, the label "Contém Fenilalanina" (Portuguese for "Contains Phenylalanine") is also mandatory in products which contain it. These warnings are placed to help individuals avoid such foods.
D-, L- and DL-phenylalanine edit
The stereoisomer D-phenylalanine (DPA) can be produced by conventional organic synthesis, either as a single enantiomer or as a component of the racemic mixture. It does not participate in protein biosynthesis although it is found in proteins in small amounts - particularly aged proteins and food proteins that have been processed. The biological functions of D-amino acids remain unclear, although D-phenylalanine has pharmacological activity at niacin receptor 2.[17]
DL-Phenylalanine (DLPA) is marketed as a nutritional supplement for its purported analgesic and antidepressant activities, which have been supported by clinical trials.[18][19][20] DL-Phenylalanine is a mixture of D-phenylalanine and L-phenylalanine. The reputed analgesic activity of DL-phenylalanine may be explained by the possible blockage by D-phenylalanine of enkephalin degradation by the enzyme carboxypeptidase A.[21][22] Enkephalins act as agonists of the mu and delta opioid receptors, and agonists of these receptors are known to produce antidepressant effects.[23] The mechanism of DL-phenylalanine's supposed antidepressant activity may also be accounted for in part by the precursor role of L-phenylalanine in the synthesis of the neurotransmitters norepinephrine and dopamine, though clinical trials have not found an antidepressant effect from L-phenylalanine alone.[18] Elevated brain levels of norepinephrine and dopamine are thought to have an antidepressant effect. D-Phenylalanine is absorbed from the small intestine and transported to the liver via the portal circulation. A small amount of D-phenylalanine appears to be converted to L-phenylalanine. D-Phenylalanine is distributed to the various tissues of the body via the systemic circulation. It appears to cross the blood–brain barrier less efficiently than L-phenylalanine, and so a small amount of an ingested dose of D-phenylalanine is excreted in the urine without penetrating the central nervous system.[24]
L-Phenylalanine is an antagonist at α2δ Ca2+ calcium channels with a Ki of 980 nM.[25]
In the brain, L-phenylalanine is a competitive antagonist at the glycine binding site of NMDA receptor[26] and at the glutamate binding site of AMPA receptor.[27] At the glycine binding site of NMDA receptor L-phenylalanine has an apparent equilibrium dissociation constant (KB) of 573 μM estimated by Schild regression[28] which is considerably lower than brain L-phenylalanine concentration observed in untreated human phenylketonuria.[29] L-Phenylalanine also inhibits neurotransmitter release at glutamatergic synapses in hippocampus and cortex with IC50 of 980 μM, a brain concentration seen in classical phenylketonuria, whereas D-phenylalanine has a significantly smaller effect.[27]
Commercial synthesis edit
L-Phenylalanine is produced for medical, feed, and nutritional applications, such as aspartame, in large quantities by utilizing the bacterium Escherichia coli, which naturally produces aromatic amino acids like phenylalanine. The quantity of L-phenylalanine produced commercially has been increased by genetically engineering E. coli, such as by altering the regulatory promoters or amplifying the number of genes controlling enzymes responsible for the synthesis of the amino acid.[30]
Derivatives edit
Boronophenylalanine (BPA) is a dihydroxyboryl derivative of phenylalanine, used in neutron capture therapy.
4-Azido-L-phenylalanine is a protein-incorporated unnatural amino acid used as a tool for bioconjugation in the field of chemical biology.
References edit
- ^ a b Ihlefeldt FS, Pettersen FB, von Bonin A, Zawadzka M, Görbitz PC (2014). "The Polymorphs of L-Phenylalanine". Angew. Chem. Int. Ed. 53 (49): 13600–13604. doi:10.1002/anie.201406886. PMID 25336255.
- ^ Dawson RM, et al. (1959). Data for Biochemical Research. Oxford: Clarendon Press.
- ^ "Nomenclature and Symbolism for Amino Acids and Peptides". IUPAC-IUB Joint Commission on Biochemical Nomenclature. 1983. Archived from the original on 9 October 2008. Retrieved 5 March 2018.
- ^ "IUPAC-IUB Commission on Biochemical Nomenclature A One-Letter Notation for Amino Acid Sequences". Journal of Biological Chemistry. 243 (13): 3557–3559. 10 July 1968. doi:10.1016/S0021-9258(19)34176-6.
- ^ Thorpe TE (1913). A Dictionary of Applied Chemistry. Longmans, Green, and Co. pp. 191–193. Retrieved 2012-06-04.
- ^ Plimmer RH (1912) [1908]. Plimmer RH, Hopkins FG (eds.). The Chemical Composition of the Proteins. Monographs on Biochemistry. Vol. Part I. Analysis (2nd ed.). London: Longmans, Green and Co. pp. 93–97. Retrieved 2012-06-04.
- ^ Ross HM, Roth J (1 April 1991). The Mood Control Diet: 21 Days to Conquering Depression and Fatigue. Simon & Schuster. p. 59. ISBN 978-0-13-590449-7.
- ^ Zeratsky K. "Phenylalanine in diet soda: Is it harmful?". Mayo Clinic. Retrieved 30 April 2019.
- ^ Institute of Medicine (2002). "Protein and Amino Acids". Dietary Reference Intakes for Energy, Carbohydrates, Fiber, Fat, Fatty Acids, Cholesterol, Protein, and Amino Acids. Washington, DC: The National Academies Press. pp. 589–768. doi:10.17226/10490. ISBN 978-0-309-08525-0.
- ^ Elango R, Ball RO, Pencharz PB (August 2012). "Recent advances in determining protein and amino acid requirements in humans". British Journal of Nutrition. 108 (S2): S22–S30. doi:10.1017/S0007114512002504. ISSN 0007-1145. PMID 23107531.
- ^ Eriksson JG, Guzzardi MA, Iozzo P, Kajantie E, Kautiainen H, Salonen MK (2017-01-01). "Higher serum phenylalanine concentration is associated with more rapid telomere shortening in men". The American Journal of Clinical Nutrition. 105 (1): 144–150. doi:10.3945/ajcn.116.130468. ISSN 0002-9165. PMID 27881392.
- ^ Broadley KJ (March 2010). "The vascular effects of trace amines and amphetamines". Pharmacology & Therapeutics. 125 (3): 363–375. doi:10.1016/j.pharmthera.2009.11.005. PMID 19948186.
- ^ Lindemann L, Hoener MC (May 2005). "A renaissance in trace amines inspired by a novel GPCR family". Trends in Pharmacological Sciences. 26 (5): 274–281. doi:10.1016/j.tips.2005.03.007. PMID 15860375.
- ^ Wang X, Li J, Dong G, Yue J (February 2014). "The endogenous substrates of brain CYP2D". European Journal of Pharmacology. 724: 211–218. doi:10.1016/j.ejphar.2013.12.025. PMID 24374199.
- ^ Nelson DL, Cox MM (2000). Lehninger, Principles of Biochemistry (3rd ed.). New York: Worth Publishing. ISBN 1-57259-153-6.
- ^ "Aspartame". UK: Food Standards Agency. Archived from the original on 2012-02-21. Retrieved 2007-06-19.
- ^ "D-Phenylalanine: Biological activity". The IUPHAR/BPS Guide to PHARMACOLOGY. Retrieved 27 December 2018.
- ^ a b Wood DR, Reimherr FW, Wender PH (1985). "Treatment of attention deficit disorder with DL-phenylalanine". Psychiatry Research. 16 (1). Elsevier BV: 21–26. doi:10.1016/0165-1781(85)90024-1. ISSN 0165-1781. PMID 3903813. S2CID 3077060.
- ^ Beckmann H, Strauss MA, Ludolph E (1977). "DL-Phenylalanine in depressed patients: An open study". Journal of Neural Transmission. 41 (2–3). Springer Science and Business Media LLC: 123–134. doi:10.1007/bf01670277. ISSN 0300-9564. PMID 335027. S2CID 5849451.
- ^ Beckmann H, Athen D, Olteanu M, Zimmer R (1979). "DL-Phenylalanine versus imipramine: A double-blind controlled study". Archiv für Psychiatrie und Nervenkrankheiten. 227 (1). Springer Science and Business Media LLC: 49–58. doi:10.1007/bf00585677. ISSN 0003-9373. PMID 387000. S2CID 23531579.
- ^ "D-Phenylalanine: Clinical data". The IUPHAR/BPS Guide to PHARMACOLOGY. Retrieved 27 December 2018.
- ^ Christianson DW, Mangani S, Shoham G, Lipscomb WN (August 1989). "Binding of D-phenylalanine and D-tyrosine to carboxypeptidase A" (PDF). The Journal of Biological Chemistry. 264 (22): 12849–12853. doi:10.1016/S0021-9258(18)51564-7. PMID 2568989.
- ^ Jelen LA, Stone JM, Young AH, Mehta MA (2022). "The opioid system in depression". Neuroscience & Biobehavioral Reviews. 140. Elsevier BV: 104800. doi:10.1016/j.neubiorev.2022.104800. ISSN 0149-7634. PMC 10166717. PMID 35914624. S2CID 251163234.
- ^ Lehmann WD, Theobald N, Fischer R, Heinrich HC (1983-03-14). "Stereospecificity of phenylalanine plasma kinetics and hydroxylation in man following oral application of a stable isotope-labelled pseudo-racemic mixture of L- and D-phenylalanine". Clinica Chimica Acta; International Journal of Clinical Chemistry. 128 (2–3): 181–198. doi:10.1016/0009-8981(83)90319-4. ISSN 0009-8981. PMID 6851137.
- ^ Mortell KH, Anderson DJ, Lynch JJ, Nelson SL, Sarris K, McDonald H, et al. (March 2006). "Structure-activity relationships of alpha-amino acid ligands for the alpha2delta subunit of voltage-gated calcium channels". Bioorganic & Medicinal Chemistry Letters. 16 (5): 1138–4111. doi:10.1016/j.bmcl.2005.11.108. PMID 16380257.
- ^ Glushakov AV, Dennis DM, Morey TE, Sumners C, Cucchiara RF, Seubert CN, et al. (2002). "Specific inhibition of N-methyl-D-aspartate receptor function in rat hippocampal neurons by L-phenylalanine at concentrations observed during phenylketonuria". Molecular Psychiatry. 7 (4): 359–367. doi:10.1038/sj.mp.4000976. PMID 11986979.
- ^ a b Glushakov AV, Dennis DM, Sumners C, Seubert CN, Martynyuk AE (April 2003). "L-Phenylalanine selectively depresses currents at glutamatergic excitatory synapses". Journal of Neuroscience Research. 72 (1): 116–124. doi:10.1002/jnr.10569. PMID 12645085. S2CID 42087834.
- ^ Glushakov AV, Glushakova O, Varshney M, Bajpai LK, Sumners C, Laipis PJ, et al. (February 2005). "Long-term changes in glutamatergic synaptic transmission in phenylketonuria". Brain. 128 (Pt 2): 300–307. doi:10.1093/brain/awh354. PMID 15634735.
- ^ Möller HE, Weglage J, Bick U, Wiedermann D, Feldmann R, Ullrich K (December 2003). "Brain imaging and proton magnetic resonance spectroscopy in patients with phenylketonuria". Pediatrics. 112 (6 Pt 2): 1580–1583. doi:10.1542/peds.112.S4.1580. hdl:11858/00-001M-0000-0010-A24A-C. PMID 14654669. S2CID 2198040.
- ^ Sprenger GA (2007). "Aromatic Amino Acids". Amino Acid Biosynthesis: Pathways, Regulation and Metabolic Engineering (1st ed.). Springer. pp. 106–113. ISBN 978-3-540-48595-7.
External links edit
- Phenylalanine mass spectrum
- Phenylalanine at ChemSynthesis