


Summary
A substitution reaction (also known as single displacement reaction or single substitution reaction) is a chemical reaction during which one functional group in a chemical compound is replaced by another functional group.[1] Substitution reactions are of prime importance in organic chemistry. Substitution reactions in organic chemistry are classified either as electrophilic or nucleophilic depending upon the reagent involved, whether a reactive intermediate involved in the reaction is a carbocation, a carbanion or a free radical, and whether the substrate is aliphatic or aromatic. Detailed understanding of a reaction type helps to predict the product outcome in a reaction. It also is helpful for optimizing a reaction with regard to variables such as temperature and choice of solvent.
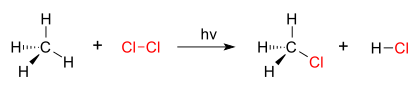
A good example of a substitution reaction is halogenation. When chlorine gas (Cl2) is irradiated, some of the molecules are split into two chlorine radicals (Cl•), whose free electrons are strongly nucleophilic. One of them breaks a C–H covalent bond in CH4 and grabs the hydrogen atom to form the electrically neutral HCl. The other radical reforms a covalent bond with the CH3• to form CH3Cl (methyl chloride).
|
|
| chlorination of methane by chlorine |
|---|
Nucleophilic substitution edit
In organic (and inorganic) chemistry, nucleophilic substitution is a fundamental class of reactions in which a nucleophile selectively bonds with or attacks the positive or partially positive charge on an atom or a group of atoms. As it does so, it replaces a weaker nucleophile, which then becomes a leaving group; the remaining positive or partially positive atom becomes an electrophile. The whole molecular entity of which the electrophile and the leaving group are part is usually called the substrate.[1]
The most general form for the reaction may be given as

where R−LG indicates the substrate. The electron pair (:) from the nucleophile (Nuc:) attacks the substrate (R−LG), forming a new covalent bond Nuc−R−LG. The prior state of charge is restored when the leaving group (LG) departs with an electron pair. The principal product in this case is R−Nuc. In such reactions, the nucleophile is usually electrically neutral or negatively charged, whereas the substrate is typically neutral or positively charged.
An example of nucleophilic substitution is the hydrolysis of an alkyl bromide, R−Br, under basic conditions, where the attacking nucleophile is the base OH− and the leaving group is Br−:

Nucleophilic substitution reactions are commonplace in organic chemistry, and they can be broadly categorized as taking place at a carbon of a saturated aliphatic compound carbon or (less often) at an aromatic or other unsaturated carbon center.[1]
Mechanisms edit
Nucleophilic substitutions can proceed by two different mechanisms, unimolecular nucleophilic substitution (SN1) and bimolecular nucleophilic substitution (SN2). The two reactions are named according tho their rate law, with SN1 having a first-order rate law, and SN2 having a second-order.[2]

The SN1 mechanism has two steps. In the first step, the leaving group departs, forming a carbocation (C+). In the second step, the nucleophilic reagent (Nuc:) attaches to the carbocation and forms a covalent sigma bond. If the substrate has a chiral carbon, this mechanism can result in either inversion of the stereochemistry or retention of configuration. Usually, both occur without preference. The result is racemization.
The stability of a carbocation (C+) depends on how many other carbon atoms are bonded to it. This results in SN1 reactions usually occurring on atoms with at least two carbons bonded to them.[2] A more detailed explanation of this can be found in the main SN1 reaction page.

The SN2 mechanism has just one step. The attack of the reagent and the expulsion of the leaving group happen simultaneously. This mechanism always results in inversion of configuration. If the substrate that is under nucleophilic attack is chiral, the reaction will therefore lead to an inversion of its stereochemistry, called a Walden inversion.
SN2 attack may occur if the backside route of attack is not sterically hindered by substituents on the substrate. Therefore, this mechanism usually occurs at an unhindered primary carbon center. If there is steric crowding on the substrate near the leaving group, such as at a tertiary carbon center, the substitution will involve an SN1 rather than an SN2.[2]

Other types of nucleophilic substitution include, nucleophilic acyl substitution, and nucleophilic aromatic substitution. Acyl substitution occurs when a nucleophile attacks a carbon that is doubly bonded to one oxygen and singly bonded to another oxygen (can be N or S or a halogen), called an acyl group. The nucleophile attacks the carbon causing the double bond to break into a single bond. The double can then reform, kicking off the leaving group in the process.
Aromatic substitution occurs on compounds with systems of double bonds connected in rings. See aromatic compounds for more.
Electrophilic substitution edit
Electrophiles are involved in electrophilic substitution reactions, particularly in electrophilic aromatic substitutions.
In this example, the benzene ring's electron resonance structure is attacked by an electrophile E+. The resonating bond is broken and a carbocation resonating structure results. Finally a proton is kicked out and a new aromatic compound is formed.

|
| 1: Free benzene + electrophile; 2a: Benzene attacks electrophile;
2b: Resonance of benzene-electrophile intermediate; 3: Substituted reaction product |
|---|
Electrophilic reactions to other unsaturated compounds than arenes generally lead to electrophilic addition rather than substitution.
Radical substitution edit
A radical substitution reaction involves radicals. An example is the Hunsdiecker reaction.
Organometallic substitution edit
Coupling reactions are a class of metal-catalyzed reactions involving an organometallic compound RM and an organic halide R′X that together react to form a compound of the type R-R′ with formation of a new carbon–carbon bond. Examples include the Heck reaction, Ullmann reaction, and Wurtz–Fittig reaction. Many variations exist.[3]
Substituted compounds edit
Substituted compounds are compounds where one or more hydrogen atoms have been replaced with something else such as an alkyl, hydroxy, or halogen. More can be found on the substituted compounds page.
Inorganic and organometallic chemistry edit
While it is common to discuss substitution reactions in the context of organic chemistry, the reaction is generic and applies to a wide range of compounds. Ligands in coordination complexes are susceptible to substitution. Both associative and dissociative mechanisms have been observed.[4][5]
Associative substitution, for example, is typically applied to organometallic and coordination complexes, but resembles the Sn2 mechanism in organic chemistry. The opposite pathway is dissociative substitution, being analogous to the Sn1 pathway.
Examples of associative mechanisms are commonly found in the chemistry of 16e square planar metal complexes, e.g. Vaska's complex and tetrachloroplatinate. The rate law is governed by the Eigen–Wilkins Mechanism.

Dissociative substitution resembles the SN1 mechanism in organic chemistry. This pathway can be well described by the cis effect, or the labilization of CO ligands in the cis position. Complexes that undergo dissociative substitution are often coordinatively saturated and often have octahedral molecular geometry. The entropy of activation is characteristically positive for these reactions, which indicates that the disorder of the reacting system increases in the rate-determining step. Dissociative pathways are characterized by a rate determining step that involves release of a ligand from the coordination sphere of the metal undergoing substitution. The concentration of the substituting nucleophile has no influence on this rate, and an intermediate of reduced coordination number can be detected. The reaction can be described with k1, k−1 and k2, which are the rate constants of their corresponding intermediate reaction steps:
![{\displaystyle {\ce {L_{\mathit {n}}M-L<=>[-\mathrm {L} ,k_{1}][+\mathrm {L} ,k_{-1}]L_{\mathit {n}}M-\Box ->[+\mathrm {L} ',k_{2}]L_{\mathit {n}}M-L'}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/e0cfb8a46601d81d661e919a1b64b91ec3113586)
Normally the rate determining step is the dissociation of L from the complex, and [L'] does not affect the rate of reaction, leading to the simple rate equation:
![{\displaystyle {\ce {Rate={{\mathit {k}}_{1}[L_{\mathit {n}}M-L]}}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/8adaf95dcf066302cf203f0dd7252129be849f03)
Further reading edit
References edit
- ^ a b c March, Jerry (1985), Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 3rd edition, New York: Wiley, ISBN 9780471854722, OCLC 642506595
- ^ a b c Bruice, Paula Yurkanis (2011). Organic Chemistry (6th ed.). 1900 E. Lake Ave., Glenview, IL 60025: Pearson Education Inc. pp. 332–365. ISBN 978-0-321-66313-9.
{{cite book}}: CS1 maint: location (link) - ^ Elschenbroich, C.; Salzer, A. (1992). Organometallics: A Concise Introduction (2nd ed.). Weinheim: Wiley-VCH. ISBN 3-527-28165-7.
- ^ Basolo, F.; Pearson, R. G. "Mechanisms of Inorganic Reactions." John Wiley and Son: New York: 1967. ISBN 0-471-05545-X
- ^ Wilkins, R. G. (1991). Kinetics and Mechanism of Reactions of Transition Metal Complexes (2nd ed.). Weinheim: VCH. ISBN 1-56081-125-0.