


Summary
Technetium is a chemical element; it has symbol Tc and atomic number 43. It is the lightest element whose isotopes are all radioactive. Technetium and promethium are the only radioactive elements whose neighbours in the sense of atomic number are both stable. All available technetium is produced as a synthetic element. Naturally occurring technetium is a spontaneous fission product in uranium ore and thorium ore (the most common source), or the product of neutron capture in molybdenum ores. This silvery gray, crystalline transition metal lies between manganese and rhenium in group 7 of the periodic table, and its chemical properties are intermediate between those of both adjacent elements. The most common naturally occurring isotope is 99Tc, in traces only.
 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Technetium | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pronunciation | /tɛkˈniːʃ(i)əm/ | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Appearance | shiny gray metal | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Mass number | [97] | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Technetium in the periodic table | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Atomic number (Z) | 43 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Group | group 7 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Period | period 5 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Block | d-block | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Electron configuration | [Kr] 4d5 5s2 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Electrons per shell | 2, 8, 18, 13, 2 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Physical properties | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Phase at STP | solid | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Melting point | 2430 K (2157 °C, 3915 °F) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Boiling point | 4538 K (4265 °C, 7709 °F) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Density (at 20° C) | 98Tc: 11.359 g/cm3 99Tc: 11.475 g/cm3[1] | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Heat of fusion | 33.29 kJ/mol | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Heat of vaporization | 585.2 kJ/mol | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Molar heat capacity | 24.27 J/(mol·K) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
Vapor pressure (extrapolated)
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Atomic properties | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Oxidation states | −1, 0, +1,[2][citation needed] +2, +3, +4, +5, +6, +7 (a strongly acidic oxide) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Electronegativity | Pauling scale: 1.9 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Ionization energies |
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Atomic radius | empirical: 136 pm | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Covalent radius | 147±7 pm | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Van der Waals radius | 205 pm | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Other properties | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Natural occurrence | from decay | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Crystal structure | hexagonal close-packed (hcp) (hP2) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Lattice constants | c = 439.90 pm (at 20 °C)[1] | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Thermal expansion | 8.175×10−6/K (at 20 °C)[1] | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Thermal conductivity | 50.6 W/(m⋅K) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Electrical resistivity | 200 nΩ⋅m (at 20 °C) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Magnetic ordering | Paramagnetic | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Molar magnetic susceptibility | +270.0×10−6 cm3/mol (298 K)[4] | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Speed of sound thin rod | 16,200 m/s (at 20 °C) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| CAS Number | 7440-26-8 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| History | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Prediction | Dmitri Mendeleev (1871) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Discovery and first isolation | Emilio Segrè and Carlo Perrier (1937) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Isotopes of technetium | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Many of technetium's properties had been predicted by Dmitri Mendeleev before it was discovered. Mendeleev noted a gap in his periodic table and gave the undiscovered element the provisional name ekamanganese (Em). In 1937, technetium became the first predominantly artificial element to be produced, hence its name (from the Greek τεχνητός, technetos, from techne, as in "craft", "art" and having the meaning of "artificial", + -ium).
One short-lived gamma ray-emitting nuclear isomer, technetium-99m, is used in nuclear medicine for a wide variety of tests, such as bone cancer diagnoses. The ground state of the nuclide technetium-99 is used as a gamma-ray-free source of beta particles. Long-lived technetium isotopes produced commercially are byproducts of the fission of uranium-235 in nuclear reactors and are extracted from nuclear fuel rods. Because even the longest-lived isotope of technetium has a relatively short half-life (4.21 million years), the 1952 detection of technetium in red giants helped to prove that stars can produce heavier elements.
History edit
Early assumptions edit
From the 1860s through 1871, early forms of the periodic table proposed by Dmitri Mendeleev contained a gap between molybdenum (element 42) and ruthenium (element 44). In 1871, Mendeleev predicted this missing element would occupy the empty place below manganese and have similar chemical properties. Mendeleev gave it the provisional name ekamanganese (from eka-, the Sanskrit word for one) because the predicted element was one place down from the known element manganese.[6]
Early misidentifications edit
Many early researchers, both before and after the periodic table was published, were eager to be the first to discover and name the missing element. Its location in the table suggested that it should be easier to find than other undiscovered elements.
| Year | Claimant | Suggested name | Actual material |
|---|---|---|---|
| 1828 | Gottfried Osann | Polinium | Iridium |
| 1845 | Heinrich Rose | Pelopium[7] | Niobium-tantalum alloy |
| 1847 | R. Hermann | Ilmenium[8] | Niobium-tantalum alloy |
| 1877 | Serge Kern | Davyum | Iridium-rhodium-iron alloy |
| 1896 | Prosper Barrière | Lucium | Yttrium |
| 1908 | Masataka Ogawa | Nipponium | Rhenium, which was the unknown dvi-manganese[9][10] |
Irreproducible results edit

German chemists Walter Noddack, Otto Berg, and Ida Tacke reported the discovery of element 75 and element 43 in 1925, and named element 43 masurium (after Masuria in eastern Prussia, now in Poland, the region where Walter Noddack's family originated).[11] This name caused significant resentment in the scientific community, because it was interpreted as referring to victories of the German army over the Russian army in the Masuria region during World War I; as the Noddacks remained in their academic positions while the Nazis were in power, suspicions and hostility against their claim for discovering element 43 continued.[12] The group bombarded columbite with a beam of electrons and deduced element 43 was present by examining X-ray emission spectrograms.[13] The wavelength of the X-rays produced is related to the atomic number by a formula derived by Henry Moseley in 1913. The team claimed to detect a faint X-ray signal at a wavelength produced by element 43. Later experimenters could not replicate the discovery, and it was dismissed as an error.[14][15] Still, in 1933, a series of articles on the discovery of elements quoted the name masurium for element 43.[16] Some more recent attempts have been made to rehabilitate the Noddacks' claims, but they are disproved by Paul Kuroda's study on the amount of technetium that could have been present in the ores they studied: it could not have exceeded 3 × 10−11 μg/kg of ore, and thus would have been undetectable by the Noddacks' methods.[12][17]
Official discovery and later history edit
The discovery of element 43 was finally confirmed in a 1937 experiment at the University of Palermo in Sicily by Carlo Perrier and Emilio Segrè.[18] In mid-1936, Segrè visited the United States, first Columbia University in New York and then the Lawrence Berkeley National Laboratory in California. He persuaded cyclotron inventor Ernest Lawrence to let him take back some discarded cyclotron parts that had become radioactive. Lawrence mailed him a molybdenum foil that had been part of the deflector in the cyclotron.[19]
Segrè enlisted his colleague Perrier to attempt to prove, through comparative chemistry, that the molybdenum activity was indeed from an element with the atomic number 43. In 1937, they succeeded in isolating the isotopes technetium-95m and technetium-97.[20][21][disputed ] University of Palermo officials wanted them to name their discovery "panormium", after the Latin name for Palermo, Panormus. In 1947[20] element 43 was named after the Greek word τεχνητός, meaning "artificial", since it was the first element to be artificially produced.[7][11] Segrè returned to Berkeley and met Glenn T. Seaborg. They isolated the metastable isotope technetium-99m, which is now used in some ten million medical diagnostic procedures annually.[22]
In 1952, the astronomer Paul W. Merrill in California detected the spectral signature of technetium (specifically wavelengths of 403.1 nm, 423.8 nm, 426.2 nm, and 429.7 nm) in light from S-type red giants.[23] The stars were near the end of their lives but were rich in the short-lived element, which indicated that it was being produced in the stars by nuclear reactions. That evidence bolstered the hypothesis that heavier elements are the product of nucleosynthesis in stars.[21] More recently, such observations provided evidence that elements are formed by neutron capture in the s-process.[24]
Since that discovery, there have been many searches in terrestrial materials for natural sources of technetium. In 1962, technetium-99 was isolated and identified in pitchblende from the Belgian Congo in very small quantities (about 0.2 ng/kg),[24] where it originates as a spontaneous fission product of uranium-238. The Oklo natural nuclear fission reactor contains evidence that significant amounts of technetium-99 were produced and have since decayed into ruthenium-99.[24]
Characteristics edit
Physical properties edit
Technetium is a silvery-gray radioactive metal with an appearance similar to platinum, commonly obtained as a gray powder.[25] The crystal structure of the bulk pure metal is hexagonal close-packed, and crystal structures of the nanodisperse pure metal are cubic. Nanodisperse technetium does not have a split NMR spectrum, while hexagonal bulk technetium has the Tc-99-NMR spectrum split in 9 satellites.[25][26] Atomic technetium has characteristic emission lines at wavelengths of 363.3 nm, 403.1 nm, 426.2 nm, 429.7 nm, and 485.3 nm.[27] The unit cell parameters of the orthorhombic Tc metal were reported when Tc is contaminated with carbon (a = 0.2805(4), b = 0.4958(8), c = 0.4474(5)·nm for Tc-C with 1.38 wt% C and a = 0.2815(4), b = 0.4963(8), c = 0.4482(5) •nm for Tc-C with 1.96 wt% C ).[28] The metal form is slightly paramagnetic, meaning its magnetic dipoles align with external magnetic fields, but will assume random orientations once the field is removed.[29] Pure, metallic, single-crystal technetium becomes a type-II superconductor at temperatures below 7.46 K.[30][a] Below this temperature, technetium has a very high magnetic penetration depth, greater than any other element except niobium.[31]
Chemical properties edit
Technetium is located in the seventh group of the periodic table, between rhenium and manganese. As predicted by the periodic law, its chemical properties are between those two elements. Of the two, technetium more closely resembles rhenium, particularly in its chemical inertness and tendency to form covalent bonds.[32] This is consistent with the tendency of period 5 elements to resemble their counterparts in period 6 more than period 4 due to the lanthanide contraction. Unlike manganese, technetium does not readily form cations (ions with a net positive charge). Technetium exhibits nine oxidation states from −1 to +7, with +4, +5, and +7 being the most common.[33] Technetium dissolves in aqua regia, nitric acid, and concentrated sulfuric acid, but it is not soluble in hydrochloric acid of any concentration.[25]
Metallic technetium slowly tarnishes in moist air[33] and, in powder form, burns in oxygen. When reacting with hydrogen at high pressure, it forms the hydride TcH1.3[34] while reacting with carbon it forms Tc6C,[35] with cell parameter 3.98 Å, as well the nanodisperce low-carbon-content carbide with parameter 4.02 Å.[36]
Technetium can catalyse the destruction of hydrazine by nitric acid, and this property is due to its multiplicity of valencies.[37] This caused a problem in the separation of plutonium from uranium in nuclear fuel processing, where hydrazine is used as a protective reductant to keep plutonium in the trivalent rather than the more stable tetravalent state. The problem was exacerbated by the mutually enhanced solvent extraction of technetium and zirconium at the previous stage,[38] and required a process modification.
Compounds edit
Pertechnetate and derivatives edit

The most prevalent form of technetium that is easily accessible is sodium pertechnetate, Na[TcO4]. The majority of this material is produced by radioactive decay from [99MoO4]2−:[39][40]
- [99MoO4]2− → [99mTcO4]− + e−
Pertechnetate (TcO−
4) is only weakly hydrated in aqueous solutions,[41] and it behaves analogously to perchlorate anion, both of which are tetrahedral. Unlike permanganate (MnO−
4), it is only a weak oxidizing agent.
Related to pertechnetate is technetium heptoxide. This pale-yellow, volatile solid is produced by oxidation of Tc metal and related precursors:
- 4 Tc + 7 O2 → 2 Tc2O7
It is a molecular metal oxide, analogous to manganese heptoxide. It adopts a centrosymmetric structure with two types of Tc−O bonds with 167 and 184 pm bond lengths.[42]
Technetium heptoxide hydrolyzes to pertechnetate and pertechnetic acid, depending on the pH:[43][44]
- Tc2O7 + 2 OH− → 2 TcO4− + H2O
- Tc2O7 + H2O → 2 HTcO4
HTcO4 is a strong acid. In concentrated sulfuric acid, [TcO4]− converts to the octahedral form TcO3(OH)(H2O)2, the conjugate base of the hypothetical triaquo complex [TcO3(H2O)3]+.[45]
Other chalcogenide derivatives edit
Technetium forms a dioxide,[46] disulfide, diselenide, and ditelluride. An ill-defined Tc2S7 forms upon treating pertechnate with hydrogen sulfide. It thermally decomposes into disulfide and elemental sulfur.[47] Similarly the dioxide can be produced by reduction of the Tc2O7.
Unlike the case for rhenium, a trioxide has not been isolated for technetium. However, TcO3 has been identified in the gas phase using mass spectrometry.[48]
Simple hydride and halide complexes edit
Technetium forms the simple complex TcH2−
9. The potassium salt is isostructural with ReH2−
9.[49] At high pressure formation of TcH1,3 from elements was also reported.[50]

The following binary (containing only two elements) technetium halides are known: TcF6, TcF5, TcCl4, TcBr4, TcBr3, α-TcCl3, β-TcCl3, TcI3, α-TcCl2, and β-TcCl2. The oxidation states range from Tc(VI) to Tc(II). Technetium halides exhibit different structure types, such as molecular octahedral complexes, extended chains, layered sheets, and metal clusters arranged in a three-dimensional network.[51][52] These compounds are produced by combining the metal and halogen or by less direct reactions.
TcCl4 is obtained by chlorination of Tc metal or Tc2O7 Upon heating, TcCl4 gives the corresponding Tc(III) and Tc(II) chlorides.[52]
- TcCl4 → α-TcCl3 + 1/2 Cl2
- TcCl3 → β-TcCl2 + 1/2 Cl2
The structure of TcCl4 is composed of infinite zigzag chains of edge-sharing TcCl6 octahedra. It is isomorphous to transition metal tetrachlorides of zirconium, hafnium, and platinum.[52]

Two polymorphs of technetium trichloride exist, α- and β-TcCl3. The α polymorph is also denoted as Tc3Cl9. It adopts a confacial bioctahedral structure.[53] It is prepared by treating the chloro-acetate Tc2(O2CCH3)4Cl2 with HCl. Like Re3Cl9, the structure of the α-polymorph consists of triangles with short M-M distances. β-TcCl3 features octahedral Tc centers, which are organized in pairs, as seen also for molybdenum trichloride. TcBr3 does not adopt the structure of either trichloride phase. Instead it has the structure of molybdenum tribromide, consisting of chains of confacial octahedra with alternating short and long Tc—Tc contacts. TcI3 has the same structure as the high temperature phase of TiI3, featuring chains of confacial octahedra with equal Tc—Tc contacts.[52]
Several anionic technetium halides are known. The binary tetrahalides can be converted to the hexahalides [TcX6]2− (X = F, Cl, Br, I), which adopt octahedral molecular geometry.[24] More reduced halides form anionic clusters with Tc–Tc bonds. The situation is similar for the related elements of Mo, W, Re. These clusters have the nuclearity Tc4, Tc6, Tc8, and Tc13. The more stable Tc6 and Tc8 clusters have prism shapes where vertical pairs of Tc atoms are connected by triple bonds and the planar atoms by single bonds. Every technetium atom makes six bonds, and the remaining valence electrons can be saturated by one axial and two bridging ligand halogen atoms such as chlorine or bromine.[54]
Coordination and organometallic complexes edit

Technetium forms a variety of coordination complexes with organic ligands. Many have been well-investigated because of their relevance to nuclear medicine.[55]
Technetium forms a variety of compounds with Tc–C bonds, i.e. organotechnetium complexes. Prominent members of this class are complexes with CO, arene, and cyclopentadienyl ligands.[56] The binary carbonyl Tc2(CO)10 is a white volatile solid.[57] In this molecule, two technetium atoms are bound to each other; each atom is surrounded by octahedra of five carbonyl ligands. The bond length between technetium atoms, 303 pm,[58][59] is significantly larger than the distance between two atoms in metallic technetium (272 pm). Similar carbonyls are formed by technetium's congeners, manganese and rhenium.[60] Interest in organotechnetium compounds has also been motivated by applications in nuclear medicine.[56] Technetium also forms aquo-carbonyl complexes, one prominent complex being [Tc(CO)3(H2O)3]+, which are unusual compared to other metal carbonyls.[56]
Isotopes edit
Technetium, with atomic number Z = 43, is the lowest-numbered element in the periodic table for which all isotopes are radioactive. The second-lightest exclusively radioactive element, promethium, has atomic number 61.[33] Atomic nuclei with an odd number of protons are less stable than those with even numbers, even when the total number of nucleons (protons + neutrons) is even,[61] and odd numbered elements have fewer stable isotopes.
The most stable radioactive isotopes are technetium-97 with a half-life of 4.21 million years, technetium-98 with 4.2 million years, and technetium-99 with 211,100 years.[62] Thirty other radioisotopes have been characterized with mass numbers ranging from 85 to 118.[63] Most of these have half-lives that are less than an hour, the exceptions being technetium-93 (2.73 hours), technetium-94 (4.88 hours), technetium-95 (20 hours), and technetium-96 (4.3 days).[64]
The primary decay mode for isotopes lighter than technetium-98 (98Tc) is electron capture, producing molybdenum (Z = 42).[63] For technetium-98 and heavier isotopes, the primary mode is beta emission (the emission of an electron or positron), producing ruthenium (Z = 44), with the exception that technetium-100 can decay both by beta emission and electron capture.[63][65]
Technetium also has numerous nuclear isomers, which are isotopes with one or more excited nucleons. Technetium-97m (97mTc; "m" stands for metastability) is the most stable, with a half-life of 91 days and excitation energy 0.0965 MeV.[64] This is followed by technetium-95m (61 days, 0.03 MeV), and technetium-99m (6.01 hours, 0.142 MeV).[64] Technetium-99m emits only gamma rays and decays to technetium-99.[64]
Technetium-99 (99Tc) is a major product of the fission of uranium-235 (235U), making it the most common and most readily available isotope of technetium. One gram of technetium-99 produces 6.2×108 disintegrations per second (in other words, the specific activity of 99Tc is 0.62 GBq/g).[29]
Occurrence and production edit
Technetium occurs naturally in the Earth's crust in minute concentrations of about 0.003 parts per trillion. Technetium is so rare because the half-lives of 97Tc and 98Tc are only 4.2 million years. More than a thousand of such periods have passed since the formation of the Earth, so the probability of survival of even one atom of primordial technetium is effectively zero. However, small amounts exist as spontaneous fission products in uranium ores. A kilogram of uranium contains an estimated 1 nanogram (10−9 g) equivalent to ten trillion atoms of technetium.[21][66][67] Some red giant stars with the spectral types S-, M-, and N contain a spectral absorption line indicating the presence of technetium.[25][68] These red giants are known informally as technetium stars.
Fission waste product edit
In contrast to the rare natural occurrence, bulk quantities of technetium-99 are produced each year from spent nuclear fuel rods, which contain various fission products. The fission of a gram of uranium-235 in nuclear reactors yields 27 mg of technetium-99, giving technetium a fission product yield of 6.1%.[29] Other fissile isotopes produce similar yields of technetium, such as 4.9% from uranium-233 and 6.21% from plutonium-239.[69] An estimated 49,000 TBq (78 metric tons) of technetium was produced in nuclear reactors between 1983 and 1994, by far the dominant source of terrestrial technetium.[70][71] Only a fraction of the production is used commercially.[b]
Technetium-99 is produced by the nuclear fission of both uranium-235 and plutonium-239. It is therefore present in radioactive waste and in the nuclear fallout of fission bomb explosions. Its decay, measured in becquerels per amount of spent fuel, is the dominant contributor to nuclear waste radioactivity after about 104 to 106 years after the creation of the nuclear waste.[70] From 1945 to 1994, an estimated 160 TBq (about 250 kg) of technetium-99 was released into the environment during atmospheric nuclear tests.[70][72] The amount of technetium-99 from nuclear reactors released into the environment up to 1986 is on the order of 1000 TBq (about 1600 kg), primarily by nuclear fuel reprocessing; most of this was discharged into the sea. Reprocessing methods have reduced emissions since then, but as of 2005 the primary release of technetium-99 into the environment is by the Sellafield plant, which released an estimated 550 TBq (about 900 kg) from 1995 to 1999 into the Irish Sea.[71] From 2000 onwards the amount has been limited by regulation to 90 TBq (about 140 kg) per year.[73] Discharge of technetium into the sea resulted in contamination of some seafood with minuscule quantities of this element. For example, European lobster and fish from west Cumbria contain about 1 Bq/kg of technetium.[74][75][c]
Fission product for commercial use edit
The metastable isotope technetium-99m is continuously produced as a fission product from the fission of uranium or plutonium in nuclear reactors:
![{\displaystyle {\ce {^{238}_{92}U ->[{\ce {sf}}] ^{137}_{53}I + ^{99}_{39}Y + 2^{1}_{0}n}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/69e432873016029d0ca91cbd92c76fc15631fd66)
![{\displaystyle {\ce {^{99}_{39}Y ->[\beta^-][1.47\,{\ce {s}}] ^{99}_{40}Zr ->[\beta^-][2.1\,{\ce {s}}] ^{99}_{41}Nb ->[\beta^-][15.0\,{\ce {s}}] ^{99}_{42}Mo ->[\beta^-][65.94\,{\ce {h}}] ^{99}_{43}Tc ->[\beta^-][211,100\,{\ce {y}}] ^{99}_{44}Ru}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/33a9d13cb741fa5c8efce11540847d7deac9654b)
Because used fuel is allowed to stand for several years before reprocessing, all molybdenum-99 and technetium-99m is decayed by the time that the fission products are separated from the major actinides in conventional nuclear reprocessing. The liquid left after plutonium–uranium extraction (PUREX) contains a high concentration of technetium as TcO−
4 but almost all of this is technetium-99, not technetium-99m.[77]
The vast majority of the technetium-99m used in medical work is produced by irradiating dedicated highly enriched uranium targets in a reactor, extracting molybdenum-99 from the targets in reprocessing facilities,[40] and recovering at the diagnostic center the technetium-99m produced upon decay of molybdenum-99.[78][79] Molybdenum-99 in the form of molybdate MoO2−
4 is adsorbed onto acid alumina (Al
2O
3) in a shielded column chromatograph inside a technetium-99m generator ("technetium cow", also occasionally called a "molybdenum cow"). Molybdenum-99 has a half-life of 67 hours, so short-lived technetium-99m (half-life: 6 hours), which results from its decay, is being constantly produced.[21] The soluble pertechnetate TcO−
4 can then be chemically extracted by elution using a saline solution. A drawback of this process is that it requires targets containing uranium-235, which are subject to the security precautions of fissile materials.[80][81]

Almost two-thirds of the world's supply comes from two reactors; the National Research Universal Reactor at Chalk River Laboratories in Ontario, Canada, and the High Flux Reactor at Nuclear Research and Consultancy Group in Petten, Netherlands. All major reactors that produce technetium-99m were built in the 1960s and are close to the end of life. The two new Canadian Multipurpose Applied Physics Lattice Experiment reactors planned and built to produce 200% of the demand of technetium-99m relieved all other producers from building their own reactors. With the cancellation of the already tested reactors in 2008, the future supply of technetium-99m became problematic.[82]
Waste disposal edit
The long half-life of technetium-99 and its potential to form anionic species creates a major concern for long-term disposal of radioactive waste. Many of the processes designed to remove fission products in reprocessing plants aim at cationic species such as caesium (e.g., caesium-137) and strontium (e.g., strontium-90). Hence the pertechnetate escapes through those processes. Current disposal options favor burial in continental, geologically stable rock. The primary danger with such practice is the likelihood that the waste will contact water, which could leach radioactive contamination into the environment. The anionic pertechnetate and iodide tend not to adsorb into the surfaces of minerals, and are likely to be washed away. By comparison plutonium, uranium, and caesium tend to bind to soil particles. Technetium could be immobilized by some environments, such as microbial activity in lake bottom sediments,[83] and the environmental chemistry of technetium is an area of active research.[84]
An alternative disposal method, transmutation, has been demonstrated at CERN for technetium-99. In this process, the technetium (technetium-99 as a metal target) is bombarded with neutrons to form the short-lived technetium-100 (half-life = 16 seconds) which decays by beta decay to stable ruthenium-100. If recovery of usable ruthenium is a goal, an extremely pure technetium target is needed; if small traces of the minor actinides such as americium and curium are present in the target, they are likely to undergo fission and form more fission products which increase the radioactivity of the irradiated target. The formation of ruthenium-106 (half-life 374 days) from the 'fresh fission' is likely to increase the activity of the final ruthenium metal, which will then require a longer cooling time after irradiation before the ruthenium can be used.[85]
The actual separation of technetium-99 from spent nuclear fuel is a long process. During fuel reprocessing, it comes out as a component of the highly radioactive waste liquid. After sitting for several years, the radioactivity reduces to a level where extraction of the long-lived isotopes, including technetium-99, becomes feasible. A series of chemical processes yields technetium-99 metal of high purity.[86]
Neutron activation edit
Molybdenum-99, which decays to form technetium-99m, can be formed by the neutron activation of molybdenum-98.[87] When needed, other technetium isotopes are not produced in significant quantities by fission, but are manufactured by neutron irradiation of parent isotopes (for example, technetium-97 can be made by neutron irradiation of ruthenium-96).[88]
Particle accelerators edit
The feasibility of technetium-99m production with the 22-MeV-proton bombardment of a molybdenum-100 target in medical cyclotrons following the reaction 100Mo(p,2n)99mTc was demonstrated in 1971.[89] The recent shortages of medical technetium-99m reignited the interest in its production by proton bombardment of isotopically enriched (>99.5%) molybdenum-100 targets.[90][91] Other techniques are being investigated for obtaining molybdenum-99 from molybdenum-100 via (n,2n) or (γ,n) reactions in particle accelerators.[92][93][94]
Applications edit
Nuclear medicine and biology edit
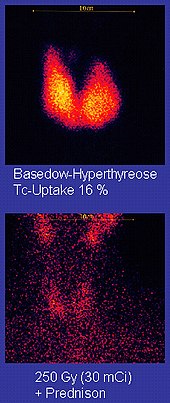
Technetium-99m ("m" indicates that this is a metastable nuclear isomer) is used in radioactive isotope medical tests. For example, Technetium-99m is a radioactive tracer that medical imaging equipment tracks in the human body.[21][90] It is well suited to the role because it emits readily detectable 140 keV gamma rays, and its half-life is 6.01 hours (meaning that about 94% of it decays to technetium-99 in 24 hours).[29] The chemistry of technetium allows it to be bound to a variety of biochemical compounds, each of which determines how it is metabolized and deposited in the body, and this single isotope can be used for a multitude of diagnostic tests. More than 50 common radiopharmaceuticals are based on technetium-99m for imaging and functional studies of the brain, heart muscle, thyroid, lungs, liver, gall bladder, kidneys, skeleton, blood, and tumors.[95]
The longer-lived isotope, technetium-95m with a half-life of 61 days, is used as a radioactive tracer to study the movement of technetium in the environment and in plant and animal systems.[96]
Industrial and chemical edit
Technetium-99 decays almost entirely by beta decay, emitting beta particles with consistent low energies and no accompanying gamma rays. Moreover, its long half-life means that this emission decreases very slowly with time. It can also be extracted to a high chemical and isotopic purity from radioactive waste. For these reasons, it is a National Institute of Standards and Technology (NIST) standard beta emitter, and is used for equipment calibration.[97] Technetium-99 has also been proposed for optoelectronic devices and nanoscale nuclear batteries.[98]
Like rhenium and palladium, technetium can serve as a catalyst. In processes such as the dehydrogenation of isopropyl alcohol, it is a far more effective catalyst than either rhenium or palladium. However, its radioactivity is a major problem in safe catalytic applications.[99]
When steel is immersed in water, adding a small concentration (55 ppm) of potassium pertechnetate(VII) to the water protects the steel from corrosion, even if the temperature is raised to 250 °C (523 K).[100] For this reason, pertechnetate has been used as an anodic corrosion inhibitor for steel, although technetium's radioactivity poses problems that limit this application to self-contained systems.[101] While (for example) CrO2−
4 can also inhibit corrosion, it requires a concentration ten times as high. In one experiment, a specimen of carbon steel was kept in an aqueous solution of pertechnetate for 20 years and was still uncorroded.[100] The mechanism by which pertechnetate prevents corrosion is not well understood, but seems to involve the reversible formation of a thin surface layer (passivation). One theory holds that the pertechnetate reacts with the steel surface to form a layer of technetium dioxide which prevents further corrosion; the same effect explains how iron powder can be used to remove pertechnetate from water. The effect disappears rapidly if the concentration of pertechnetate falls below the minimum concentration or if too high a concentration of other ions is added.[102]
As noted, the radioactive nature of technetium (3 MBq/L at the concentrations required) makes this corrosion protection impractical in almost all situations. Nevertheless, corrosion protection by pertechnetate ions was proposed (but never adopted) for use in boiling water reactors.[102]
Precautions edit
Technetium plays no natural biological role and is not normally found in the human body.[25] Technetium is produced in quantity by nuclear fission, and spreads more readily than many radionuclides. It appears to have low chemical toxicity. For example, no significant change in blood formula, body and organ weights, and food consumption could be detected for rats which ingested up to 15 µg of technetium-99 per gram of food for several weeks.[103] In the body, technetium quickly gets converted to the stable TcO−
4 ion, which is highly water-soluble and quickly excreted. The radiological toxicity of technetium (per unit of mass) is a function of compound, type of radiation for the isotope in question, and the isotope's half-life.[104]
All isotopes of technetium must be handled carefully. The most common isotope, technetium-99, is a weak beta emitter; such radiation is stopped by the walls of laboratory glassware. The primary hazard when working with technetium is inhalation of dust; such radioactive contamination in the lungs can pose a significant cancer risk. For most work, careful handling in a fume hood is sufficient, and a glove box is not needed.[105]
Notes edit
- ^ Irregular crystals and trace impurities raise this transition temperature to 11.2 K for 99.9% pure technetium powder.[30]
- ^ As of 2005[update], technetium-99 in the form of ammonium pertechnetate is available to holders of an Oak Ridge National Laboratory permit.[25]
- ^ The anaerobic, spore-forming bacteria in the Clostridium genus are able to reduce Tc(VII) to Tc(IV). Clostridia bacteria play a role in reducing iron, manganese, and uranium, thereby affecting these elements' solubility in soil and sediments. Their ability to reduce technetium may determine a large part of mobility of technetium in industrial wastes and other subsurface environments.[76]
References edit
- ^ a b c Arblaster, John W. (2018). Selected Values of the Crystallographic Properties of Elements. Materials Park, Ohio: ASM International. ISBN 978-1-62708-155-9.
- ^ "Technetium: technetium(III) iodide compound data". OpenMOPAC.net. Retrieved 2007-12-10.
- ^ Mattolat, C.; Gottwald, T.; Raeder, S.; Rothe, S.; Schwellnus, F.; Wendt, K.; Thörle-Pospiech, P.; Trautmann, N. (24 May 2010). "Determination of the first ionization potential of technetium". Physical Review A. 81: 052513. doi:10.1103/PhysRevA.81.052513.
- ^ Weast, Robert (1984). CRC, Handbook of Chemistry and Physics. Boca Raton, Florida: Chemical Rubber Company Publishing. pp. E110. ISBN 0-8493-0464-4.
- ^ Kondev, F. G.; Wang, M.; Huang, W. J.; Naimi, S.; Audi, G. (2021). "The NUBASE2020 evaluation of nuclear properties" (PDF). Chinese Physics C. 45 (3): 030001. doi:10.1088/1674-1137/abddae.
- ^ Jonge; Pauwels, E. K. (1996). "Technetium, the missing element". European Journal of Nuclear Medicine. 23 (3): 336–44. doi:10.1007/BF00837634. PMID 8599967. S2CID 24026249.
- ^ a b Holden, N. E. "History of the Origin of the Chemical Elements and Their Discoverers". Brookhaven National Laboratory. Retrieved 2009-05-05.
- ^ Hermann, R. (1847). "Untersuchungen über das Ilmenium". Journal für Praktische Chemie. 40: 457–480. doi:10.1002/prac.184704001110.
- ^ Yoshihara, H. K. (2004). "Discovery of a new element 'nipponium': re-evaluation of pioneering works of Masataka Ogawa and his son Eijiro Ogawa". Spectrochimica Acta Part B. 59 (8): 1305–1310. Bibcode:2004AcSpe..59.1305Y. doi:10.1016/j.sab.2003.12.027.
- ^ Hisamatsu, Yoji; Egashira, Kazuhiro; Maeno, Yoshiteru (2022). "Ogawa's nipponium and its re-assignment to rhenium". Foundations of Chemistry. 24: 15–57. doi:10.1007/s10698-021-09410-x. Retrieved 16 November 2023.
- ^ a b van der Krogt, P. "Technetium". Elentymolgy and Elements Multidict. Retrieved 2009-05-05.
- ^ a b Eric Scerri, A tale of seven elements, (Oxford University Press 2013) ISBN 978-0-19-539131-2, pp. 109–114, 125–131
- ^ Emsley 2001, p. 423.
- ^ Armstrong, J. T. (2003). "Technetium". Chemical & Engineering News. 81 (36): 110. doi:10.1021/cen-v081n036.p110. Retrieved 2009-11-11.
- ^ Nies, K. A. (2001). "Ida Tacke and the warfare behind the discovery of fission". Archived from the original on 2009-08-09. Retrieved 2009-05-05.
- ^ Weeks, M. E. (1933). "The discovery of the elements. XX. Recently discovered elements". Journal of Chemical Education. 10 (3): 161–170. Bibcode:1933JChEd..10..161W. doi:10.1021/ed010p161.
- ^ Habashi, Fathi (2006). "The History of Element 43—Technetium". Journal of Chemical Education. 83 (2): 213. Bibcode:2006JChEd..83..213H. doi:10.1021/ed083p213.1. Retrieved 2 January 2023.
- ^ Heiserman, D. L. (1992). "Element 43: Technetium". Exploring Chemical Elements and their Compounds. New York: TAB Books. p. 164. ISBN 978-0-8306-3018-9.
- ^ Segrè, Emilio (1993). A Mind Always in Motion: The Autobiography of Emilio Segrè. Berkeley, California: University of California Press. pp. 115–118. ISBN 978-0520076273.
- ^ a b Perrier, C.; Segrè, E. (1947). "Technetium: The Element of Atomic Number 43". Nature. 159 (4027): 24. Bibcode:1947Natur.159...24P. doi:10.1038/159024a0. PMID 20279068. S2CID 4136886.
- ^ a b c d e Emsley 2001, pp. 422–425
- ^ Hoffman, Darleane C.; Ghiorso, Albert; Seaborg, Glenn T. (2000). "Chapter 1.2: Early Days at the Berkeley Radiation Laboratory". The Transuranium People: The Inside Story. University of California, Berkeley & Lawrence Berkeley National Laboratory. p. 15. ISBN 978-1-86094-087-3. Archived from the original on 2007-01-24. Retrieved 2007-03-31.
- ^ Merrill, P. W. (1952). "Technetium in the stars". Science. 115 (2992): 479–489 [484]. Bibcode:1952Sci...115..479.. doi:10.1126/science.115.2992.479. PMID 17792758.
- ^ a b c d Schwochau 2000, pp. 7–9
- ^ a b c d e f Hammond 2004, p. [page needed].
- ^ Tarasov, V.P.; Muravlev, Yu. B.; German, K. E.; Popova, N. N. (2001). "99Tc NMR of Supported Technetium Nanoparticles". Doklady Physical Chemistry. 377 (1–3): 71–76. doi:10.1023/A:1018872000032. S2CID 91522281.
- ^ Lide, David R. (2004–2005). "Line Spectra of the Elements". The CRC Handbook. CRC press. pp. 10–70 (1672). ISBN 978-0-8493-0595-5.
- ^ German, K. E.; Peretrukhin, V. F.; Gedgovd, K. N.; Grigoriev, M. S.; Tarasov, A. V.; Plekhanov, Yu V.; Maslennikov, A. G.; Bulatov, G. S.; Tarasov, V. P.; Lecomte, M. (2005). "Tc Carbide and New Orthorhombic Tc Metal Phase". Journal of Nuclear and Radiochemical Sciences. 6 (3): 211–214. doi:10.14494/jnrs2000.6.3_211.
- ^ a b c d Rimshaw, S. J. (1968). Hampel, C. A. (ed.). The Encyclopedia of the Chemical Elements. New York: Reinhold Book Corporation. pp. 689–693.
- ^ a b Schwochau 2000, p. 96.
- ^ Autler, S. H. (1968). "Technetium as a Material for AC Superconductivity Applications" (PDF). Proceedings of the 1968 Summer Study on Superconducting Devices and Accelerators. Retrieved 2009-05-05.
- ^ Greenwood & Earnshaw 1997, p. 1044.
- ^ a b c Husted, R. (2003-12-15). "Technetium". Periodic Table of the Elements. Los Alamos National Laboratory. Retrieved 2009-10-11.
- ^ Zhou, Di; Semenok, Dmitrii V.; Volkov, Mikhail A.; Troyan, Ivan A.; Seregin, Alexey Yu.; Chepkasov, Ilya V.; Sannikov, Denis A.; Lagoudakis, Pavlos G.; Oganov, Artem R.; German, Konstantin E. (2023-02-06). "Synthesis of technetium hydride $\mathrm{Tc}{\mathrm{H}}_{1.3}$ at 27 GPa". Physical Review B. 107 (6): 064102. arXiv:2210.01518. doi:10.1103/PhysRevB.107.064102.
- ^ German, K. E.; Peretrukhin, V. F.; Gedgovd, K. N.; Grigoriev, M. S.; Tarasov, A. V.; Plekhanov, Yu V.; Maslennikov, A. G.; Bulatov, G. S.; Tarasov, V. P.; Lecomte, M. (2005). "Tc Carbide and New Orthorhombic Tc Metal Phase". Journal of Nuclear and Radiochemical Sciences. 6 (3): 211–214. doi:10.14494/jnrs2000.6.3_211.
- ^ Kuznetsov, Vitaly V.; German, Konstantin E.; Nagovitsyna, Olga A.; Filatova, Elena A.; Volkov, Mikhail A.; Sitanskaia, Anastasiia V.; Pshenichkina, Tatiana V. (2023-10-31). "Route to Stabilization of Nanotechnetium in an Amorphous Carbon Matrix: Preparative Methods, XAFS Evidence, and Electrochemical Studies". Inorganic Chemistry. doi:10.1021/acs.inorgchem.3c03001. ISSN 0020-1669.
- ^ Garraway, John (1984). "The technetium-catalysed oxidation of hydrazine by nitric acid". Journal of the Less Common Metals. 97: 191–203. doi:10.1016/0022-5088(84)90023-7.
- ^ Garraway, J. (1985). "Coextraction of pertechnetate and zirconium by tri-n-butyl phosphate". Journal of the Less Common Metals. 106 (1): 183–192. doi:10.1016/0022-5088(85)90379-0.
- ^ Schwochau 2000, pp. 127–136.
- ^ a b Moore, P. W. (April 1984). "Technetium-99 in generator systems" (PDF). Journal of Nuclear Medicine. 25 (4): 499–502. PMID 6100549. Retrieved 2012-05-11.
- ^ Ustynyuk, Yuri A.; Gloriozov, Igor P.; Zhokhova, Nelly I.; German, Konstantin E.; Kalmykov, Stepan N. (2021-11-15). "Hydration of the pertechnetate anion. DFT study". Journal of Molecular Liquids. 342: 117404. doi:10.1016/j.molliq.2021.117404. ISSN 0167-7322.
- ^ Krebs, B. (1969). "Technetium(VII)-oxid: Ein Übergangsmetalloxid mit Molekülstruktur im festen Zustand (Technetium(VII) Oxide, a Transition Metal Oxide with a Molecular Structure in the Solid State)". Angewandte Chemie. 81 (9): 328–329. doi:10.1002/ange.19690810905.
- ^ Schwochau 2000, p. 127.
- ^ Herrell, A. Y.; Busey, R. H.; Gayer, K. H. (1977). Technetium(VII) Oxide, in Inorganic Syntheses. Vol. XVII. pp. 155–158. ISBN 978-0-07-044327-3.
- ^ Poineau F; Weck PF; German K; Maruk A; Kirakosyan G; Lukens W; Rego DB; et al. (2010). "Speciation of heptavalent technetium in sulfuric acid: structural and spectroscopic studies" (PDF). Dalton Transactions. 39 (37): 8616–8619. doi:10.1039/C0DT00695E. PMID 20730190. S2CID 9419843. Archived from the original (PDF) on 2017-03-05. Retrieved 2011-11-14.
- ^ Schwochau 2000, p. 108.
- ^ Schwochau 2000, pp. 112–113.
- ^ Gibson, John K. (1993). "High-Temperature Oxide and Hydroxide Vapor Species of Technetium". Radiochimica Acta. 60 (2–3): 121–126. doi:10.1524/ract.1993.60.23.121. S2CID 99795348.
- ^ Schwochau 2000, p. 146.
- ^ Zhou, Di; Semenok, Dmitrii V.; Volkov, Mikhail A.; Troyan, Ivan A.; Seregin, Alexey Yu.; Chepkasov, Ilya V.; Sannikov, Denis A.; Lagoudakis, Pavlos G.; Oganov, Artem R.; German, Konstantin E. (2023-02-06). "Synthesis of technetium hydride $\mathrm{Tc}{\mathrm{H}}_{1.3}$ at 27 GPa". Physical Review B. 107 (6): 064102. arXiv:2210.01518. doi:10.1103/PhysRevB.107.064102.
- ^ Johnstone, E. V. (May 2014). Binary Technetium Halides. UNLV Theses, Dissertations, Professional Papers, and Capstones (Thesis). University of Nevada, Las Vegas. doi:10.34917/5836118.
- ^ a b c d Poineau, Frederic; Johnstone, Erik V.; Czerwinski, Kenneth R.; Sattelberger, Alfred P. (2014). "Recent Advances in Technetium Halide Chemistry". Accounts of Chemical Research. 47 (2): 624–632. doi:10.1021/ar400225b. PMID 24393028.
- ^ Poineau, Frederic; Johnstone, Erik V.; Weck, Philippe F.; Kim, Eunja; Forster, Paul M.; Scott, Brian L.; Sattelberger, Alfred P.; Czerwinski, Kenneth R. (2010). "Synthesis and Structure of Technetium Trichloride". Journal of the American Chemical Society. 132 (45): 15864–5. doi:10.1021/ja105730e. PMID 20977207.
- ^ German, K. E.; Kryutchkov, S. V. (2002). "Polynuclear Technetium Halide Clusters". Russian Journal of Inorganic Chemistry. 47 (4): 578–583. Archived from the original on 2015-12-22.
- ^ Bartholomä, Mark D.; Louie, Anika S.; Valliant, John F.; Zubieta, Jon (2010). "Technetium and Gallium Derived Radiopharmaceuticals: Comparing and Contrasting the Chemistry of Two Important Radiometals for the Molecular Imaging Era". Chemical Reviews. 110 (5): 2903–20. doi:10.1021/cr1000755. PMID 20415476.
- ^ a b c Alberto, Roger (2010). "Organometallic Radiopharmaceuticals". Medicinal Organometallic Chemistry. Topics in Organometallic Chemistry. Vol. 32. pp. 219–246. doi:10.1007/978-3-642-13185-1_9. ISBN 978-3-642-13184-4.
- ^ Hileman, J. C.; Huggins, D. K.; Kaesz, H. D. (1961). "Technetium carbonyl". Journal of the American Chemical Society. 83 (13): 2953–2954. doi:10.1021/ja01474a038.
- ^ Bailey, M. F.; Dahl, Lawrence F. (1965). "The Crystal Structure of Ditechnetium Decacarbonyl". Inorganic Chemistry. 4 (8): 1140–1145. doi:10.1021/ic50030a011.
- ^ Wallach, D. (1962). "Unit cell and space group of technetium carbonyl, Tc2(CO)10". Acta Crystallographica. 15 (10): 1058. doi:10.1107/S0365110X62002789.
- ^ Schwochau 2000, pp. 286, 328.
- ^ Clayton, D. D. (1983). Principles of stellar evolution and nucleosynthesis: with a new preface. University of Chicago Press. p. 547. ISBN 978-0-226-10953-4.
- ^ Audi, G.; Kondev, F. G.; Wang, M.; Huang, W. J.; Naimi, S. (2017). "The NUBASE2016 evaluation of nuclear properties" (PDF). Chinese Physics C. 41 (3): 030001. Bibcode:2017ChPhC..41c0001A. doi:10.1088/1674-1137/41/3/030001.
- ^ a b c Sonzogni, A. A. (ed.). "Chart of Nuclides". New York: National Nuclear Data Center, Brookhaven National Laboratory. Archived from the original on 2009-08-25. Retrieved 2009-11-11.
- ^ a b c d Holden, N. E. (2006). Lide. D. R. (ed.). Handbook of Chemistry and Physics (87th ed.). Boca Raton, Florida: CRC Press. pp. 11-88–11-89. ISBN 978-0-8493-0487-3.
- ^ Lide, David R., ed. (2004–2005). "Table of the isotopes". The CRC Handbook of Chemistry and Physics. CRC press.
- ^ Dixon, P.; Curtis, David B.; Musgrave, John; Roensch, Fred; Roach, Jeff; Rokop, Don (1997). "Analysis of Naturally Produced Technetium and Plutonium in Geologic Materials". Analytical Chemistry. 69 (9): 1692–1699. doi:10.1021/ac961159q. PMID 21639292.
- ^ Curtis, D.; Fabryka-Martin, June; Dixon, Paul; Cramer, Jan (1999). "Nature's uncommon elements: plutonium and technetium". Geochimica et Cosmochimica Acta. 63 (2): 275. Bibcode:1999GeCoA..63..275C. doi:10.1016/S0016-7037(98)00282-8.
- ^ Moore, C. E. (1951). "Technetium in the Sun". Science. 114 (2951): 59–61. Bibcode:1951Sci...114...59M. doi:10.1126/science.114.2951.59. PMID 17782983.
- ^ Schwochau 2000, pp. 374–404.
- ^ a b c Yoshihara, K. (1996). "Technetium in the Environment". In Yoshihara, K.; Omori, T. (eds.). Technetium and Rhenium: Their Chemistry and Its Applications. Topics in Current Chemistry. Vol. 176. Berlin, Heidelberg: Springer-Verlag. pp. 17–35. doi:10.1007/3-540-59469-8_2. ISBN 978-3-540-59469-7.
- ^ a b Garcia-Leon, M. (2005). "99Tc in the Environment: Sources, Distribution and Methods" (PDF). Journal of Nuclear and Radiochemical Sciences. 6 (3): 253–259. doi:10.14494/jnrs2000.6.3_253.
- ^ Desmet, G.; Myttenaere, C. (1986). Technetium in the environment. Springer. p. 69. ISBN 978-0-85334-421-6.
- ^ Tagami, K. (2003). "Technetium-99 Behaviour in the Terrestrial Environment — Field Observations and Radiotracer Experiments". Journal of Nuclear and Radiochemical Sciences. 4: A1–A8. doi:10.14494/jnrs2000.4.a1.
- ^ Szefer, P.; Nriagu, J. O. (2006). Mineral components in foods. CRC Press. p. 403. ISBN 978-0-8493-2234-1.
- ^ Harrison, J. D.; Phipps, A. (2001). "Gut transfer and doses from environmental technetium". J. Radiol. Prot. 21 (1): 9–11. Bibcode:2001JRP....21....9H. doi:10.1088/0952-4746/21/1/004. PMID 11281541. S2CID 250752077.
- ^ Francis, A. J.; Dodge, C. J.; Meinken, G. E. (2002). "Biotransformation of pertechnetate by Clostridia". Radiochimica Acta. 90 (9–11): 791–797. doi:10.1524/ract.2002.90.9-11_2002.791. S2CID 83759112.
- ^ Schwochau 2000, p. 39.
- ^ US 3799883, Hirofumi Arino, "Silver coated charcoal step", issued March 26, 1974, assigned to Union Carbide Corporation
- ^ Committee on Medical Isotope Production Without Highly Enriched Uranium (2009). Medical Isotope Production Without Highly Enriched Uranium. National Academies Press. p. vii. ISBN 978-0-309-13040-0.
- ^ Lützenkirchen, K.-R. "Nuclear forensics sleuths trace the origin of trafficked material". Los Alamos National Laboratory. Archived from the original on 2013-02-16. Retrieved 2009-11-11.
- ^ Snelgrove, J. L.; Hofman, G. L. (1995). Development and Processing of LEU Targets for Mo-99 Production (PDF). 1995 International Meeting on Reduced Enrichment for Research and Test Reactors, September 18–21, 1994, Paris, France. ANL.gov. Retrieved 2009-05-05.
- ^ Thomas, Gregory S.; Maddahi, Jamshid (2010). "The technetium shortage". Journal of Nuclear Cardiology. 17 (6): 993–8. doi:10.1007/s12350-010-9281-8. PMID 20717761. S2CID 2397919.
- ^ German, Konstantin E.; Firsova, E. V.; Peretrukhin, V. F.; Khizhnyak, T. V.; Simonoff, M. (2003). "Bioaccumulation of Tc, Pu, and Np on Bottom Sediments in Two Types of Freshwater Lakes of the Moscow Oblast". Radiochemistry. 45 (6): 250–6. doi:10.1023/A:1026008108860. S2CID 55030255.
- ^ Shaw, G. (2007). Radioactivity in the terrestrial environment. Elsevier. p. 147. ISBN 978-0-08-043872-6.
- ^ Altomare, P; Bernardi (1979). Alternative disposal concepts for high-level and transuranic radioactive waste disposal. US Environmental Protection Agency.
- ^ Schwochau 2000, pp. 87–96.
- ^ "Manual for reactor produced radioisotopes" (PDF). IAEA. January 2003. Retrieved 2009-08-27.
- ^ Kelly, J. J. (1980). Effluent and environmental radiation surveillance: a symposium. ASTM International. p. 91.
- ^ Beaver, J. E.; Hupf, H.B. (November 1971). "Production of 99mTc on a Medical Cyclotron: a Feasibility Study" (PDF). Journal of Nuclear Medicine. 12 (11): 739–741. PMID 5113635.
- ^ a b Laurence Knight (30 May 2015). "The element that can make bones glow". BBC. Retrieved 30 May 2015.
- ^ Guérin B; Tremblay S; Rodrigue S; Rousseau JA; et al. (2010). "Cyclotron production of 99mTc: an approach to the medical isotope crisis" (PDF). Journal of Nuclear Medicine. 51 (4): 13N–6N. PMID 20351346.
- ^ Scholten, Bernhard; Lambrecht, Richard M.; Cogneau, Michel; Vera Ruiz, Hernan; Qaim, Syed M. (25 May 1999). "Excitation functions for the cyclotron production of 99mTc and 99Mo". Applied Radiation and Isotopes. 51 (1): 69–80. doi:10.1016/S0969-8043(98)00153-5.
- ^ Takács, S.; Szűcs, Z.; Tárkányi, F.; Hermanne, A.; Sonck, M. (1 January 2003). "Evaluation of proton induced reactions on 100Mo: New cross sections for production of 99mTc and 99Mo". Journal of Radioanalytical and Nuclear Chemistry. 257 (1): 195–201. doi:10.1023/A:1024790520036. S2CID 93040978.
- ^ Celler, A.; Hou, X.; Bénard, F.; Ruth, T. (2011). "Theoretical modeling of yields for proton-induced reactions on natural and enriched molybdenum targets". Physics in Medicine and Biology. 56 (17): 5469–5484. Bibcode:2011PMB....56.5469C. doi:10.1088/0031-9155/56/17/002. PMID 21813960. S2CID 24231457.
- ^ Schwochau 2000, p. 414.
- ^ Schwochau 2000, pp. 12–27.
- ^ Schwochau 2000, p. 87.
- ^ James S. Tulenko; Dean Schoenfeld; David Hintenlang; Carl Crane; Shannon Ridgeway; Jose Santiago; Charles Scheer (2006-11-30). University Research Program in Robotics REPORT (PDF) (Report). University of Florida. doi:10.2172/895620. Retrieved 2007-10-12.
- ^ Schwochau 2000, pp. 87–90.
- ^ a b Emsley 2001, p. 425.
- ^ "Ch. 14 Separation Techniques" (PDF). EPA: 402-b-04-001b-14-final. US Environmental Protection Agency. July 2004. Archived (PDF) from the original on 2014-03-08. Retrieved 2008-08-04.
- ^ a b Schwochau 2000, p. 91.
- ^ Desmet, G.; Myttenaere, C. (1986). Technetium in the environment. Springer. pp. 392–395. ISBN 978-0-85334-421-6.
- ^ Schwochau 2000, pp. 371–381.
- ^ Schwochau 2000, p. 40.
Bibliography edit
- Emsley, J. (2001). Nature's Building Blocks: An A-Z Guide to the Elements. Oxford, England, UK: Oxford University Press. ISBN 978-0-19-850340-8.
- Greenwood, N. N.; Earnshaw, A. (1997). Chemistry of the Elements (2nd ed.). Oxford: Butterworth-Heinemann. ISBN 978-0-7506-3365-9.
- Hammond, C. R. (2004). "The Elements". Handbook of Chemistry and Physics (81st ed.). CRC press. ISBN 978-0-8493-0485-9.
- Scerri, Eric (2013). A Tale of Seven Elements. Oxford University Press, ISBN 9780195391312.
- Schwochau, K. (2000). Technetium: Chemistry and Radiopharmaceutical Applications. Weinheim, Germany: Wiley-VCH. ISBN 978-3-527-29496-1.
Further reading edit
- Choppin, G.; Liljenzin, J.-O.; Rydberg, J. (2002). "Nuclear Mass and Stability". Radiochemistry and nuclear chemistry (3rd ed.). Butterworth-Heinemann. pp. 41–57. ISBN 978-0-7506-7463-8.
- Cotton, F. A.; Wilkinson, G.; Murillo, C. A.; Bochmann, M. (1999). Advanced Inorganic Chemistry (6th ed.). New York: John Wiley & Sons, Inc. ISBN 978-0-471-19957-1.
- Scerri, E. R. (2007). The Periodic Table, Its Story and Its Significance. Oxford University Press. ISBN 978-0-19-530573-9.
- Wilson, B.J., ed. (1966). The Radiochemical Manual (2nd ed.). AEA Technology. ISBN 978-0-7058-1768-4.
- EnvironmentalChemistry.com – Technetium
- Nudat 2 Archived 2021-04-28 at the Wayback Machine nuclide chart from the National Nuclear Data Center, Brookhaven National Laboratory
External links edit
- Technetium at The Periodic Table of Videos (University of Nottingham)