


Summary
Thin-film solar cells are made by depositing one or more thin layers (thin films or TFs) of photovoltaic material onto a substrate, such as glass, plastic or metal. Thin-film solar cells are typically a few nanometers (nm) to a few microns (µm) thick–much thinner than the wafers used in conventional crystalline silicon (c-Si) based solar cells, which can be up to 200 µm thick. Thin-film solar cells are commercially used in several technologies, including cadmium telluride (CdTe), copper indium gallium diselenide (CIGS), and amorphous thin-film silicon (a-Si, TF-Si).





- Top: thin-film silicon laminates being installed onto a roof.
- Middle: CIGS solar cell on a flexible plastic backing and rigid CdTe panels mounted on a supporting structure
- Bottom: thin-film laminates on rooftops
Solar cells are often classified into so-called generations based on the active (sunlight-absorbing) layers used to produce them, with the most well-established or first-generation solar cells being made of single- or multi-crystalline silicon. This is the dominant technology currently used in most solar PV systems. Most thin-film solar cells are classified as second generation, made using thin layers of well-studied materials like amorphous silicon (a-Si), cadmium telluride (CdTe), copper indium gallium selenide (CIGS), or gallium arsenide (GaAs). Solar cells made with newer, less established materials are classified as third-generation or emerging solar cells. This includes some innovative thin-film technologies, such as perovskite, dye-sensitized, quantum dot, organic, and CZTS thin-film solar cells.
Thin-film cells have several advantages over first-generation silicon solar cells, including being lighter and more flexible due to their thin construction. This makes them suitable for use in building-integrated photovoltaics and as semi-transparent, photovoltaic glazing material that can be laminated onto windows. Other commercial applications use rigid thin film solar panels (interleaved between two panes of glass) in some of the world's largest photovoltaic power stations. Additionally, the materials used in thin-film solar cells are typically produced using simple and scalable methods more cost-effective than first-generation cells, leading to lower environmental impacts like greenhouse gas (GHG) emissions in many cases. Thin-film cells also typically outperform renewable and non-renewable sources for electricity generation in terms of human toxicity and heavy-metal emissions.
Despite initial challenges with efficient light conversion, especially among third-generation PV materials, as of 2023 some thin-film solar cells have reached efficiencies of up to 29.1% for single-junction thin-film GaAs cells, exceeding the maximum of 26.1% efficiency for standard single-junction first-generation solar cells. Multi-junction concentrator cells incorporating thin-film technologies have reached efficiencies of up to 47.6% as of 2023.[1]
Still, many thin-film technologies have been found to have shorter operational lifetimes and larger degradation rates than first-generation cells in accelerated life testing, which has contributed to their somewhat limited deployment. Globally, the PV marketshare of thin-film technologies remains around 5% as of 2023.[2] However, thin-film technology has become considerably more popular in the United States, where CdTe cells alone accounted for nearly 30% of new utility-scale deployment in 2022.[3]
History edit
Early research into thin-film solar cells began in the 1970s. In 1970, Zhores Alferov's team at Ioffe Institute created the first gallium arsenide (GaAs) solar cells, later winning the 2000 Nobel prize in Physics for this and other work.[4][5] Two years later in 1972, Prof. Karl Böer founded the Institute of Energy Conversion (IEC) at the University of Delaware to further thin-film solar research. The institute first focused on copper sulfide/cadmium sulfide (Cu2S/CdS) cells and later expanded to zinc phosphide (Zn3P2) and amorphous silicon (a-Si) thin-films as well in 1975.[6] In 1973, the IEC debuted a solar-powered house, Solar One, in the first example of residential building-integrated photovoltaics.[7] In the next decade, interest in thin-film technology for commercial use and aerospace[8] applications increased significantly, with several companies beginning development of amorphous silicon thin-film solar devices.[9] Thin-film solar efficiencies rose to 10% for Cu2S/CdS in 1980,[10] and in 1986 ARCO Solar launched the first commercially-available thin-film solar cell, the G-4000, made from amorphous silicon.[11]
In the 1990s and 2000s, thin-film solar cells saw significant increases in maximum efficiencies and expansion of existing thin-film technologies into new sectors. In 1992, a thin-film solar cell with greater than 15% efficiency was developed at University of South Florida.[12] Only seven years later in 1999, the U.S. National Renewable Energy Laboratory (NREL) and Spectrolab collaborated on a three-junction gallium arsenide solar cell that reached 32% efficiency.[13] That same year, Kiss + Cathcart designed transparent thin-film solar cells for some of the windows in 4 Times Square, generating enough electricity to power 5-7 houses.[14][12] In 2000, BP Solar introduced two new commercial solar cells based on thin-film technology.[12] In 2001, the first organic thin-film solar cells were developed at the Johannes Kepler University of Linz. In 2005, GaAs solar cells got even thinner with the first free-standing (no substrate) cells introduced by researchers at Radboud University.[15]
This was also a time of significant advances in the exploration of new third-generation solar materials–materials with the potential to overcome theoretical efficiency limits for traditional solid-state materials.[16] In 1991, the first high-efficiency dye-sensitized solar cell was developed, replacing the ordinary solid semiconducting (active) layer of the cell with a liquid electrolyte mixture containing light-absorbing dye.[17] In the early 2000s, development of quantum dot solar cells began,[16] technology later certified by NREL in 2011.[18] In 2009, researchers at the University of Tokyo reported a new type solar cell using perovskites as the active layer and achieving over 3% efficiency,[19] building on Murase Chikao's 1999 work which created a perovskite layer capable of absorbing light.[20]
In the 2010s and early 2020s, innovation in thin-film solar technology has included efforts to expand third-generation solar technology to new applications and to decrease production costs, as well as significant efficiency improvements for both second and third generation materials. In 2015, Kyung-In Synthetic released the first inkjet solar cells, flexible solar cells made with industrial printers.[21] In 2016, Vladimir Bulović's Organic and Nanostructured Electronics (ONE) Lab at the Massachusetts Institute of Technology (MIT) created thin-film cells light enough to sit on top of soap bubbles.[22] In 2022, the same group introduced flexible organic thin-film solar cells integrated into fabric.[23][24]
Thin-film solar technology captured a peak global market share of 32% of the new photovoltaic deployment in 1988 before declining for several decades and reaching another, smaller peak of 17% again in 2009.[25][26] Market share then steadily declined to 5% in 2021 globally,[25] however thin-film technology captured approximately 19% of the total U.S. market share in the same year, including 30% of utility-scale production.[27]
Theory of operation edit
In a typical solar cell, the photovoltaic effect is used to generate electricity from sunlight. The light-absorbing or "active layer" of the solar cell is typically a semiconducting material, meaning that there is a gap in its energy spectrum between the valence band of localized electrons around host ions and the conduction band of higher-energy electrons which are free to move throughout the material. For most semiconducting materials at room temperature, electrons which have not gained extra energy from another source will exist largely in the valence band, with few or no electrons in the conduction band. When a solar photon reaches the semiconducting active layer in a solar cell, electrons in the valence band can absorb the energy of the photon and be excited into the conduction band, allowing them to move freely throughout the material. When this happens, an empty electron state (or hole) is left behind in the valence band. Together, the conduction band electron and the valence band hole are called an electron-hole pair. Both the electron and the hole in the electron-hole pair can move freely throughout the material as electricity.[28] However, if the electron-hole pair is not separated, the electron and hole can recombine into the lower-energy original state, releasing a photon of the corresponding energy. In thermodynamic equilibrium, the forward process (absorbing a photon to excite an electron-hole pair) and reverse process (emitting a photon to destroy an electron-hole pair) must occur at the same rate by the principle of detailed balance. Therefore, to construct a solar cell from a semiconducting material and extract current during the excitation process, the electron and hole of the electron-hole pair must be separated. This can be achieved in a variety of different ways, but the most common is with a p-n junction, where a positively doped (p-type) semiconducting layer and a negatively doped (n-type) semiconducting layer meet, creating a chemical potential difference which draws electrons one direction and holes the other, separating the electron-hole pair.[29] This may instead be achieved using metal contacts with different work functions, as in a Schottky-junction cell.
In a thin-film solar cell, the process is largely the same but the active semiconducting layer is made much thinner. This may be made possible by some intrinsic property of the semiconducting material used that allows it to convert a particularly large number of photons per thickness. For example, some thin-film materials having a direct bandgap, meaning the conduction and valence band electron states are at the same momentum instead of different momenta as in the case of an indirect bandgap semiconductor like silicon. Having a direct bandgap eliminates the need for a source or sink of momentum (typically a lattice vibration, or phonon), simplifying the two-step process of absorbing a photon into a single-step process.[30] Other thin-film materials may be able to absorb more photons per thickness simply due to having an energy bandgap that is well-matched to the peak energy of the solar spectrum, meaning there are many solar photons of the correct energy available to excite electron-hole pairs.
In other thin-film solar cells, the semiconducting layer may be replaced entirely with another light-absorbing material, for example an electrolyte solution and photo-active dye molecules in a dye-sensitized solar cell or by quantum dots in a quantum dot solar cell.
Materials edit
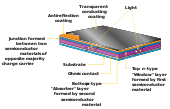
Thin-film technologies reduce the amount of active material in a cell. The active layer may be placed on a rigid substrate made from glass, plastic, or metal or the cell may be made with a flexible substrate like cloth. Thin-film solar cells tend to be cheaper than crystalline silicon cells and have a smaller ecological impact (determined from life cycle analysis).[31] Their thin and flexible nature also makes them ideal for applications like building-integrated photovoltaics. The majority of film panels have 2-3 percentage points lower conversion efficiencies than crystalline silicon,[32] though some thin-film materials outperform crystalline silicon panels in terms of efficiency. Cadmium telluride (CdTe), copper indium gallium selenide (CIGS) and amorphous silicon (a-Si) are three of the most prominent thin-film technologies.
Second-generation thin-film materials edit
Cadmium telluride edit
Cadmium telluride (CdTe) is a chalcogenide material that is the predominant thin film technology. With about 5 percent of worldwide PV production, it accounts for more than half of the thin film market. The cell's lab efficiency has also increased significantly in recent years and is on a par with CIGS thin film and close to the efficiency of multi-crystalline silicon as of 2013.[33]: 24–25 Also, CdTe has the lowest energy payback time of all mass-produced PV technologies, and can be as short as eight months in favorable locations.[33]: 31 CdTe also performs better than most other thin-film PV materials across many important environmental impact factors like global warming potential and heavy metal emissions.[34] A prominent manufacturer is the US-company First Solar based in Tempe, Arizona, that produces CdTe-panels with an efficiency of about 18 percent.[35]
Although the toxicity of cadmium may not be that much of an issue and environmental concerns completely resolved with the recycling of CdTe modules at the end of their life time,[36] there are still uncertainties[37] and the public opinion is skeptical towards this technology.[38][39] The usage of rare materials may also become a limiting factor to the industrial scalability of CdTe thin film technology. The rarity of tellurium—of which telluride is the anionic form—is comparable to that of platinum in the earth's crust and contributes significantly to the module's cost.[40]
Copper indium gallium selenide (CIGS) edit
| Cell | Module | |
| Glass substrate | 23.6 %[41] | 20.3 %[42] |
| Flexible substrate | 22.2 %[43] | 18.6 %[44] |
Like CdTe, copper indium gallium selenide (CIGS) and its variations are chalcogenide compound semiconductors. CIGS solar cells reached a lab-efficiency above 23 percent (see table) and a share of 0.8 percent in the overall PV market in 2021.[45] Numerous companies have produced CIGS solar cells and modules, however, some of them have significantly reduced or ceased production during the last years.
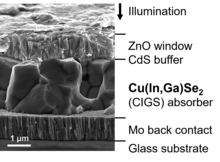
Actual research aims at improving properties related to fabrication and functionality by modifying or replacing the individual layers, for example:
- Alternative materials are investigated to replace cadmium sulfide as a buffer layer due to its hazard potential.[46]
- Molybdenium as a back contact is replaced by a transparent conducting film in order to obtain bifacial solar cells.[47]

- CIGS solar cells show a particularly low loss in efficiency, when deposited on a flexible substrate instead of glass (see table), hence they are considered promising candidates for bendable and lightweight solar modules.[48]
Apart from the development potential of the other layers in the solar cell, the absorber material CIGS has the remarkable property, that its band gap can be tuned by adjusting the ratio of indium and gallium in the compound. By tuning the band gap, the fraction of the solar spectrum which is absorbed by the solar cell can be changed, making CIGS cells especially interesting as constituents of multi-junction solar cells.[48]

It is also possible to partially replace copper by silver and selenium by sulfur yielding the compound (AgzCu1-z)(In1-xGax)(Se1-ySy)2. In order to distinguish the sulfur-free compound, it is sometimes abbrievated CIGSe, while the acronym CIGS can refer to both sulfur and selenium containing compounds. The silver containing compound is sometimes referred to as ACIGS. Variations of the CIGS composition are subject to current research and in part also fabricated in industry.
Silicon edit

There are three prominent silicon thin-film architectures:
- Amorphous silicon cells
- Amorphous / microcrystalline tandem cells (micromorph)
- Thin-film polycrystalline silicon on glass.[49]
Amorphous silicon edit
Amorphous silicon (a-Si) is a non-crystalline, allotropic form of silicon and the most well-developed thin film technology to-date. Thin-film silicon is an alternative to conventional wafer (or bulk) crystalline silicon. While chalcogenide-based CdTe and CIS thin films cells have been developed in the lab with great success, there is still industry interest in silicon-based thin film cells. Silicon-based devices exhibit fewer problems than their CdTe and CIS counterparts such as toxicity and humidity issues with CdTe cells and low manufacturing yields of CIS due to material complexity. Additionally, due to political resistance to the use non-"green" materials in solar energy production, there is no stigma in the use of standard silicon.
This type of thin-film cell is mostly fabricated by a technique called plasma-enhanced chemical vapor deposition. It uses a gaseous mixture of silane (SiH4) and hydrogen to deposit a very thin layer of only 1 micrometre (µm) of silicon on a substrate, such as glass, plastic or metal, that has already been coated with a layer of transparent conducting oxide. Other methods used to deposit amorphous silicon on a substrate include sputtering and hot wire chemical vapor deposition techniques.[50]
a-Si is attractive as a solar cell material because it's an abundant, non-toxic material. It requires a low processing temperature and enables a scalable production upon a flexible, low-cost substrate with little silicon material required. Due to its bandgap of 1.7 eV, amorphous silicon also absorbs a very broad range of the light spectrum, that includes infrared and even some ultraviolet and performs very well at weak light. This allows the cell to generate power in the early morning, or late afternoon and on cloudy and rainy days, contrary to crystalline silicon cells, that are significantly less efficient when exposed at diffuse and indirect daylight.[citation needed]
However, the efficiency of an a-Si cell suffers a significant drop of about 10 to 30 percent during the first six months of operation. This is called the Staebler-Wronski effect (SWE) – a typical loss in electrical output due to changes in photoconductivity and dark conductivity caused by prolonged exposure to sunlight. Although this degradation is perfectly reversible upon annealing at or above 150 °C, conventional c-Si solar cells do not exhibit this effect in the first place.

Its basic electronic structure is the p-i-n junction. The amorphous structure of a-Si implies high inherent disorder and dangling bonds, making it a bad conductor for charge carriers. These dangling bonds act as recombination centers that severely reduce carrier lifetime. A p-i-n structure is usually used, as opposed to an n-i-p structure. This is because the mobility of electrons in a-Si:H is roughly 1 or 2 orders of magnitude larger than that of holes, and thus the collection rate of electrons moving from the n- to p-type contact is better than holes moving from p- to n-type contact. Therefore, the p-type layer should be placed at the top where the light intensity is stronger, so that the majority of the charge carriers crossing the junction are electrons.[51]
Tandem-cell using a-Si/μc-Si edit
A layer of amorphous silicon can be combined with layers of other allotropic forms of silicon to produce a multi-junction solar cell. When only two layers (two p-n junctions) are combined, it is called a tandem-cell. By stacking these layers on top of one other, a broader range of the light spectra is absorbed, improving the cell's overall efficiency.
In micromorphous silicon, a layer of microcrystalline silicon (μc-Si) is combined with amorphous silicon, creating a tandem cell. The top a-Si layer absorbs the visible light, leaving the infrared part to the bottom μc-Si layer. The micromorph stacked-cell concept was pioneered and patented at the Institute of Microtechnology (IMT) of the Neuchâtel University in Switzerland,[52] and was licensed to TEL Solar. A new world record PV module based on the micromorph concept with 12.24% module efficiency was independently certified in July 2014.[53]
Because all layers are made of silicon, they can be manufactured using PECVD. The band gap of a-Si is 1.7 eV and that of c-Si is 1.1 eV. The c-Si layer can absorb red and infrared light. The best efficiency can be achieved at transition between a-Si and c-Si. As nanocrystalline silicon (nc-Si) has about the same bandgap as c-Si, nc-Si can replace c-Si.[54]

Tandem-cell using a-Si/pc-Si edit
Amorphous silicon can also be combined with protocrystalline silicon (pc-Si) into a tandem-cell. Protocrystalline silicon with a low volume fraction of nanocrystalline silicon is optimal for high open-circuit voltage.[55] These types of silicon present dangling and twisted bonds, which results in deep defects (energy levels in the bandgap) as well as deformation of the valence and conduction bands (band tails).
Polycrystalline silicon on glass edit
A new attempt to fuse the advantages of bulk silicon with those of thin-film devices is thin film polycrystalline silicon on glass. These modules are produced by depositing an antireflection coating and doped silicon onto textured glass substrates using plasma-enhanced chemical vapor deposition (PECVD). The texture in the glass enhances the efficiency of the cell by approximately 3% by reducing the amount of incident light reflecting from the solar cell and trapping light inside the solar cell. The silicon film is crystallized by an annealing step, temperatures of 400–600 Celsius, resulting in polycrystalline silicon.
These new devices show energy conversion efficiencies of 8% and high manufacturing yields of >90%. Crystalline silicon on glass (CSG), where the polycrystalline silicon is 1–2 micrometres, is noted for its stability and durability; the use of thin film techniques also contributes to a cost savings over bulk photovoltaics. These modules do not require the presence of a transparent conducting oxide layer. This simplifies the production process twofold; not only can this step be skipped, but the absence of this layer makes the process of constructing a contact scheme much simpler. Both of these simplifications further reduce the cost of production. Despite the numerous advantages over alternative design, production cost estimations on a per unit area basis show that these devices are comparable in cost to single-junction amorphous thin film cells.[49]
Gallium arsenide edit
Gallium arsenide (GaAs) is a III-V direct bandgap semiconductor and is a very common material used for single-crystalline thin-film solar cells. GaAs solar cells have continued to be one of the highest performing thin-film solar cells due to their exceptional heat resistant properties and high efficiencies.[56] As of 2019, single-crystalline GaAs cells have shown the highest solar cell efficiency of any single-junction solar cell with an efficiency of 29.1%.[57] This record-holding cell achieved this high efficiency by implementing a back mirror on the rear surface to increase photon absorption which allowed the cell to attain an impressive short-circuit current density and an open-circuit voltage value near the Shockley–Queisser limit.[58] As a result, GaAs solar cells have nearly reached their maximum efficiency although improvements can still be made by employing light trapping strategies.[59]
GaAs thin-films are most commonly fabricated using epitaxial growth of the semiconductor on a substrate material. The epitaxial lift-off (ELO) technique, first demonstrated in 1978, has proven to be the most promising and effective. In this method, the thin film layer is peeled off of the substrate by selectively etching a sacrificial layer that was placed between the epitaxial film and substrate.[60] The GaAs film and the substrate remain minimally damaged through the separation process, allowing for the reuse of the host substrate.[61] With reuse of the substrate the fabrication costs can be reduced, but not completely forgone, since the substrate can only be reused a limited number of times.[59] This process is still relatively costly and research is still being done to find more cost-effective ways of growing the epitaxial film layer onto a substrate.
Despite the high performance of GaAs thin-film cells, the expensive material costs hinder their ability for wide-scale adoption in the solar cell industry. GaAs is more commonly used in multi-junction solar cells for solar panels on spacecraft, as the larger power to weight ratio lowers the launch costs in space-based solar power (InGaP/(In)GaAs/Ge cells). They are also used in concentrator photovoltaics, an emerging technology best suited for locations that receive much sunlight, using lenses to focus sunlight on a much smaller, thus less expensive GaAs concentrator solar cell.

Third-generation (emerging) thin-film materials edit
The National Renewable Energy Laboratory classifies a number of thin-film technologies as emerging photovoltaics—most of them have not yet been commercially applied and are still in the research or development phase. Many use organic materials, often organometallic compounds as well as inorganic substances. Though many of these technologies have struggled with instability and low efficiencies in their early stages, some emerging materials like perovskites have been able to attain efficiencies comparable to mono crystalline silicon cells. Many of these technologies have the potential to beat the Shockley–Queisser limit for efficiency of a single-junction solid-state cell. Significant research has been invested into these technologies as they promise to achieve the goal of producing low-cost, high-efficiency solar cells with smaller environmental impacts.
Copper zinc tin sulfide (CZTS) edit
Copper zinc tin sulfide or Cu(Zn,Sn)(S,Se)2, commonly abbreviated CZTS, and its derivatives CZTSe and CZTSSe belong to a group chalcogenides (like CdTe and CIGS/CIS) sometimes called kesterites. Unlike CdTe and CIGS, CZTS is made from abundant and non-toxic raw materials. Additionally, the bandgap of CZTS can be tuned by changing the S/Se ratio, which is a desirable property for engineering of optimal solar cells.[62] CZTS also has a high light absorption coefficient.
Other emerging chalcogenide PV materials include antimony-based compounds like Sb2(S,Se)3. Like CZTS, they have tunable bandgaps and good light absorption. Antimony-based compounds also have a quasi-1D structure which may be useful for device engineering. All of these emerging chalcogenide materials have the advantage of being a part of one of the most mature and efficient families of thin-film technology. As of 2022, CZTS cells have achieved a maximum efficiency of around 12.6% while antimony-based cells have reached 9.9%.[63]
Dye-sensitized (DSPV) edit
Dye-sensitized cells, also known as Grätzel cells or DSPV, are innovative cells that perform a kind of artificial photosynthesis,[64] removing the need for a bulk solid-state semiconductor or a p-n junction. Instead, they are constructed using a layer of photoactive dye mixed with semiconductor transition metal oxide nanoparticles on top of a liquid electrolyte solution, surrounded by electrical contacts made of platinum or sometimes graphene and encapsulated in glass. When photons enter the cell, they can be absorbed by the dye molecules, putting them into their sensitized state. In this state, the dye molecules can inject electrons into the semiconductor conduction band. The dye electrons are then replenished by the electrode, preventing recombination of the electron-hole pair. The electron in the semiconductor flows out as current through the electrical contacts.[65]
Dye-sensitized solar cells are attractive because they allow for cheap and cost-efficient roll-based manufacturing.[62] In practice, however, the inclusion of expensive materials like platinum and ruthenium keep these low costs from being achieved.[62] Dye-sensitized cells also have issues with stability and degradation, particularly because of the liquid electrolyte. In high temperature environments, the electrolyte may leak from the cell while in low temperature environments the electrolyte may freeze. Some of these issues can be overcome using a quasi-solid state electrolyte.[64]
As of 2023, the maximum realized efficiency of a dye-sensitized solar cell is around 13%.[66]
Organic photovoltaics (OPV) edit
Organic solar cells use organic semiconducting polymers as the photoactive material. These organic polymers are cost-effective to produce and are tunable with high absorption coefficients.[64] Organic solar cell manufacturing is also cost effective and can make use of efficient roll-to-roll production techniques. They also have some of the lowest environmental impact scores of all PV technologies across a wide range of impact factors including energy payback time global warming potential.[67]
Organic cells and are naturally flexible, lending themselves well to many applications. Scientists at the Massachusetts Institute of Technology (MIT)'s Organic and Nanostructured Electronics Lab (ONE Lab) have integrated organic PV onto flexible fabric substrates that can be unrolled over 500 times without degradation.[23]
However, organic solar cells are generally not very stable and tend to have low operational lifetimes. They also tend to be less efficient than other thin-film cells due to some intrinsic limits of the material like a large binding energy for electron-hole pairs.[64] As of 2023, the maximum achieved efficiency for organic solar cells is 18.2%.[66]
Perovskite solar cells edit
Perovskites are a group of materials with a shared crystal structure, named after their discoverer, mineralogist Lev Perovski. The perovskites most often used for PV applications are organic-inorganic hybrid methylammonium lead halides, which host a number of advantageous properties including widely tunable bandgaps, high absorption coefficients, and good electronic transport properties for both electrons and holes.[68] As of 2023, single-junction perovskite solar cells achieved a maximum efficiency of 25.7%, rivaling that of mono crystalline silicon. Perovskites are also commonly used in tandem and multi-junction cells with crystalline silicon, CIGS, and other PV technologies to achieve even higher efficiencies.[66] They also offer a wide spectrum of low-cost applications.[69][70][71]
However, perovskite cells tend to have short lifetimes, with 5 years being a typical lifetime as of 2016.[68] This is mostly due to their chemical instability when exposed to light, moisture, UV radiation, and high temperatures which may even cause them to undergo a structural transition that impacts the operation of the device. Therefore, proper encapsulation is very important.[64]
Quantum dot photovoltaics (QDPV) edit
Quantum dot photovoltaics (QDPV) replace the usual solid-state semiconducting active layer with semiconductor quantum dots. The bandgap of the photo-active layer can be tuned by changing the size of the quantum dots.[62] QDPV has the potential to generate more than one electron-hole pair per photon in a process called multiple exciton generation (MEG) which could allow for a theoretical maximum conversion efficiency of 87%,[34] though as of 2023 the maximum achieved efficiency of a QDPV cell is around 18.1%.[66] QDPV cells also tend to use much less of the active layer material than other solar cell types leading to a low-cost manufacturing process. However, QDPV cells tend to have high environmental impacts compared to other thin-film PV materials, especially human toxicity and heavy metal emissions.[34]
Applications edit
Transparent solar cells edit
In 2022, semitransparent solar cells that are as large as windows were reported,[72] after team members of the study achieved record efficiency with high transparency in 2020.[73][74] Also in 2022, other researchers reported the fabrication of solar cells with a record average visible transparency of 79%, being nearly invisible.[75][76]
Building-integrated photovoltaics edit
Thin-film PV materials tend to be lightweight and flexible in nature, which lends itself naturally to building-integrated photovoltaics (BIPV).[77] Common examples include the integration of semi-transparent modules can be integrated into window designs[78] and the use of rigid thin-film panels to replace roofing material. BIPV can greatly reduce the lifetime environmental impacts (like greenhouse gas (GHG) emission) due to solar cell modules due to the avoided emissions associated with not utilizing the usual building materials.[79]
Efficiencies edit
Despite initially lower efficiencies at the time of their introduction, many thin-film technologies have efficiencies comparable to conventional single-junction non-concentrator crystalline silicon solar cells which have a 26.1% maximum efficiency as of 2023. Im fact, both GaAs thin-film and GaAs single-crystal cells have larger maximum efficiencies of 29.1% and 27.4% respectively. The maximum efficiencies for single-junction non-concentrator thin-film cells of various prominent thin-film materials are shown in the chart.
| Solar Cell Type | Best Efficiency (%) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| GaAs thin-film | 29.1
| ||||||||
| GaAs single crystal | 27.8
| ||||||||
| Single-crystal silicon* | 26.1
| ||||||||
| Perovskites | 25.7
| ||||||||
| Multi-crystalline silicon* | 24.4
| ||||||||
| CIGS | 23.6
| ||||||||
| CdTe | 22.1
| ||||||||
| Thin-film c-Si | 21.7
| ||||||||
| Organic | 18.2
| ||||||||
| Quantum dot | 18.1
| ||||||||
| Amorphous silicon | 14
| ||||||||
| Dye-sensitized | 13
| ||||||||
| CZTSSe | 13
| ||||||||
|
*Not thin-film, included for comparison only. Data from NREL 2023 Best Research-Cell Efficiency dataset. | |||||||||
Commercial module efficiences edit
It's important to note that the maximum efficiencies achieved in a laboratory setting are generally higher than the efficiencies of manufactured cells, which often have efficiencies 20-50% lower.[68] As of 2021, the maximum efficiency of manufactured solar cells was 24.4% for mono crystalline silicon, 20.4% for poly crystalline silicon, 12.3% for amorphous silicon, 19.2% for CIGS, and 19% for CdTe modules.[80] The thin film cell prototype with the best efficiency yields 20.4% (First Solar), comparable to the best conventional solar cell prototype efficiency of 25.6% from Panasonic.[81][82]
A previous record for thin film solar cell efficiency of 22.3% was achieved by Solar Frontier, the world's largest CIS (copper indium selenium) solar energy provider. In joint research with the New Energy and Industrial Technology Development Organization (NEDO) of Japan, Solar Frontier achieved 22.3% conversion efficiency on a 0.5 cm2 cell using its CIS technology. This was an increase of 0.6 percentage points over the industry's previous thin-film record of 21.7%.[83]
Calculation of efficiency edit

The efficiency of a solar cell quantifies the percentage of incident light on the solar cell that is converted into usable electricity. There are many factors that affect the efficiency of a solar cell, so the efficiency may be further parametrized by additional numerical quantities including the short-circuit current, open-circuit voltage, maximum power point, fill factor, and quantum efficiency. The short-circuit current is the maximum current the cell can flow with no voltage load. Similarly, the open-circuit voltage is the voltage across the device with no current or, alternatively, the voltage required for no current to flow. On a current vs. voltage (IV) curve, the open-circuit voltage is the horizontal intercept of the curve with the voltage axis and the short-circuit current is the vertical intercept of the curve with the current axis. The maximum power point is the point along the curve where the maximum power output of the solar cell is achieved and the area of the rectangle with side lengths equal to the current and voltage coordinates of the maximum power point is called the fill factor. The fill factor is a measure of how much power the solar cell achieves at this maximum power point. Intuitively, IV curves with a more square shape and a flatter top and side will have a larger fill factor and therefore a higher efficiency.[84] Whereas these parameters characterize the efficiency of the solar cell based mostly on its macroscopic electrical properties, the quantum efficiency measures either the ratio of the number of photons incident on the cell to the number of charge carriers extracted (external quantum efficiency) or the ratio of the number of photons absorbed by the cell to the number of charge carriers extracted (internal quantum efficiency). Either way, the quantum efficiency is a more direct probe of the microscopic structure of the solar cell.[85]

Increasing efficiency edit
Some third-generation solar cells boost efficiency through the integration of concentrator and/or multi-junction device geometry.[63] This can lead to efficiencies larger than the Shockley–Queisser limit of approximately 42% efficiency for a single-junction semiconductor solar cell under one-sun illumination.[86]
A multi-junction cell is one that incorporates multiple semiconducting active layers with different bandgaps. In a typical solar cell, a single absorber with a bandgap near the peak of the solar spectrum is used, and any photons with energy greater than or equal to the bandgap can excite valence-band electrons into the conduction band to create electron-hole pairs. However, any excess energy above the Fermi energy will be quickly dissipated due to thermalization, leading to voltage losses from the inability to efficiently extract the energy of high-energy photons. Mutli-junction cells are able to recoup some of this energy lost to thermalization by stacking multiple absorber layers on top of each other with the top layer absorbing the highest-energy photons and letting the lower energy photons pass through to the lower layers with smaller bandgaps, and so on. This not only allows the cells to capture energy from photons in a larger range of energies, but also extracts more energy per photon from the higher-energy photons.[87]
Concentrator photovoltaics use an optical system of lenses that sit on top of the cell to focus light from a larger area onto the device, similar to a funnel for sunlight. In addition to creating more electron-hole pairs simply by increasing the number of photons available for absorption, having a higher concentration of charge carriers can increase the efficiency of the solar cell by increasing the conductivity. The addition of a concentrator to a solar cell can not only increase efficiency, but can also reduce the space, materials, and cost needed to produce the cell.[88]
Both of these techniques are employed in the highest-efficiency solar cell as of 2023, which is a four-junction concentrator cell with 47.6% efficiency.[1]
| Solar Cell Type | Best Efficiency (%) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 4+ junction concentrator | 47.6(GaInP/AlGaAs/GaInAsP/GaInAs)
| ||||||||
| 3-junction concentrator | 44.4(GaInP/GaAs/GaInAs)
| ||||||||
| 3-junction non-concentrator | 39.46(GaInP/mQW-GaAs/GaInAs)
| ||||||||
| 4+ junction non-concentrator | 39.2(AlGaInP/AlGaAs/GaAs/GaInAs)
| ||||||||
| 2-junction concentrator | 35.5(GaInAsP/GaInAs)
| ||||||||
| 2-junction non-concentrator | 32.9(GaInP/GaAs)
| ||||||||
| Perovskite/Si tandem | 32.5
| ||||||||
| GaAs concentrator | 30.8
| ||||||||
| Si single-crystal concentrator | 27.6
| ||||||||
| Si HIT | 26.81
| ||||||||
| Perovskite/CIGS tandem | 24.2
| ||||||||
| CIGS concentrator | 23.3
| ||||||||
| Organic tandem | 14.2(PDTB-EF-T/IT-4F)
| ||||||||
|
Data from the NREL 2023 Best Research-Cell Efficiency dataset. | |||||||||
Increasing absorption edit
Multiple techniques have been employed to increase the amount of light that enters the cell and reduce the amount that escapes without absorption. The most obvious technique is to minimize the top contact coverage of the cell surface, reducing the area that blocks light from reaching the cell.
The weakly absorbed long wavelength light can be obliquely coupled into silicon and traverses the film several times to enhance absorption.[89][90]
Multiple methods have been developed to increase absorption by reducing the number of incident photons being reflected away from the cell surface. An additional anti-reflective coating can cause destructive interference within the cell by modulating the refractive index of the surface coating. Destructive interference eliminates the reflective wave, causing all incident light to enter the cell.
Surface texturing is another option for increasing absorption, but increases costs. By applying a texture to the active material's surface, the reflected light can be refracted into striking the surface again, thus reducing reflectance. For example, black silicon texturing by reactive ion etching (RIE) is an effective and economic approach to increase the absorption of thin-film silicon solar cells.[91] A textured back reflector can prevent light from escaping through the rear of the cell. Instead of applying the texturing on the active materials, photonic micro-structured coatings applied on the cells' front contact can be an interesting alternative for light-trapping, as they allow both geometric anti-reflection and light scattering while avoiding the roughening of the photovoltaic layers (thereby preventing increase of recombination).[92][93]
Besides surface texturing, the plasmonic light-trapping scheme attracted a lot of attention to aid photocurrent enhancement in thin film solar cells.[94][95] This method makes use of collective oscillation of excited free electrons in noble metal nanoparticles, which are influenced by particle shape, size and dielectric properties of the surrounding medium. Applying noble-metal nanoparticles at the back of thin-film solar cells leads to the formation of plasmonic back reflectors, which allow broadband photocurrent enhancement.[96] This is a result of both light scattering of the weakly-absorbed photons from the rear-located nanoparticles, plus improved light incoupling (geometric anti-reflection) caused by the hemispherical corrugations at the cells’ front surface formed from the conformal deposition of the cell materials over the particles.[97]
In addition to minimizing reflective loss, the solar cell material itself can be optimized to have higher chance of absorbing a photon that reaches it. Thermal processing techniques can significantly enhance the crystal quality of silicon cells and thereby increase efficiency.[98] Layering thin-film cells to create a multi-junction solar cell can also be done. Each layer's band gap can be designed to best absorb a different range of wavelengths, such that together they can absorb a greater spectrum of light.[99]
Further advancement into geometric considerations can exploit nanomaterial dimensionality. Large, parallel nanowire arrays enable long absorption lengths along the length of the wire while maintaining short minority carrier diffusion lengths along the radial direction.[100] Adding nanoparticles between the nanowires allows conduction. The natural geometry of these arrays forms a textured surface that traps more light.
Production, cost and market edit
With the advances in conventional crystalline silicon (c-Si) technology in recent years, and the falling cost of the polysilicon feedstock, that followed after a period of severe global shortage, pressure increased on manufacturers of commercial thin-film technologies, including amorphous thin-film silicon (a-Si), cadmium telluride (CdTe), and copper indium gallium diselenide (CIGS), leading to the bankruptcy of several companies.[101] As of 2013, thin-film manufacturers continue to face price competition from Chinese refiners of silicon and manufacturers of conventional c-Si solar panels. Some companies together with their patents were sold to Chinese firms below cost.[102]
edit
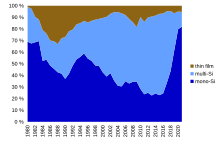
In 2013 thin-film technologies accounted for about 9 percent of worldwide deployment, while 91 percent was held by crystalline silicon (mono-Si and multi-Si). With 5 percent of the overall market, CdTe holds more than half of the thin-film market, leaving 2 percent to each CIGS and amorphous silicon.[33]: 18–19
CIGS technology edit
Several prominent manufacturers couldn't stand the pressure caused by advances in conventional c-Si technology of recent years. The company Solyndra ceased all business activity and filed for Chapter 11 bankruptcy in 2011, and Nanosolar, also a CIGS manufacturer, closed its doors in 2013. Although both companies produced CIGS solar cells, it has been pointed out, that the failure was not due to the technology but rather because of the companies themselves, using a flawed architecture, such as, for example, Solyndra's cylindrical substrates.[103] In 2014, Korean LG Electronics terminated research on CIGS restructuring its solar business, and Samsung SDI decided to cease CIGS-production, while Chinese PV manufacturer Hanergy is expected to ramp up production capacity of their 15.5% efficient, 650 mm×1650 mm CIGS-modules.[104][105] One of the largest producers of CI(G)S photovoltaics is the Japanese company Solar Frontier with a manufacturing capacity in the gigawatt-scale.[106] (Also see List of CIGS companies).
CdTe technology edit
The company First Solar, a leading manufacturer of CdTe, has been building several of the world's largest solar power stations, such as the Desert Sunlight Solar Farm and Topaz Solar Farm, both in the Californian desert with a 550 MW capacity each, as well as the 102-megawatt Nyngan Solar Plant in Australia, the largest PV power station in the Southern Hemisphere, commissioned in 2015.[107]
In 2011, GE announced plans to spend $600 million on a new CdTe solar cell plant and enter this market,[108] and in 2013, First Solar bought GE's CdTe thin-film intellectual property portfolio and formed a business partnership.[109] In 2012 Abound Solar, a manufacturer of cadmium telluride modules, went bankrupt.[110]
a-Si technology edit
In 2012, ECD solar, once one of the world's leading manufacturer of amorphous silicon (a-Si) technology, filed for bankruptcy in Michigan, United States. Swiss OC Oerlikon divested its solar division that produced a-Si/μc-Si tandem cells to Tokyo Electron Limited.[111][112]
Other companies that left the amorphous silicon thin-film market include DuPont, BP, Flexcell, Inventux, Pramac, Schuco, Sencera, EPV Solar,[113] NovaSolar (formerly OptiSolar)[114] and Suntech Power that stopped manufacturing a-Si modules in 2010 to focus on conventional silicon solar panels. In 2013, Suntech filed for bankruptcy in China.[115][116] In August 2013, the spot market price of thin-film a-Si and a-Si/µ-Si dropped to €0.36 and €0.46, respectively[117] (about $0.50 and $0.60) per watt.[118]
Thin film solar on metal roofs edit


With the increasing efficiencies of thin film solar, installing them on standing seam metal roofs has become cost competitive with traditional Monocrystalline and Polycrystalline solar cells. The thin film panels are flexible and run down the standing seam metal roofs and stick to the metal roof with Adhesive, so no holes are needed to install. The connection wires run under the ridge cap at the top of the roof. Efficiency ranges from 10-18% but only costs about $2.00-$3.00 per watt of installed capacity, compared to Monocrystalline which is 17-22% efficient and costs $3.00-$3.50 per watt of installed capacity. Thin film solar is light weight at 7-10 ounces per square foot. Thin film solar panels last 10–20 years[119] but have a quicker ROI than traditional solar panels, the metal roofs last 40–70 years before replacement compared to 12–20 years for an asphalt shingle roof.[120][121]
| Type[121] | Cost per Watt | Efficiency | Average 6 kW System Cost |
|---|---|---|---|
| Polycrystalline | $2.80-$3.00 | 13-17% | $17,400 |
| Monocrystalline | $3.00-$3.50 | 17-22% | $19,000 |
| Thin film Panels | $2.00-$3.00 | 10-18% | $17,000 |
Cost edit
In 1998, scientists at the National Renewable Energy Laboratory (NREL) predicted that production of thin-film PV systems at a cost of $50 per m2 could someday be possible, which would make them extremely economically viable. At this price, thin-film PV systems would yield return on investment of 30% or greater.[122]
To help achieve this goal, in 2022 NREL began administering the Cadmium Telluride Accelerator Consortium (CTAC) with the objective of enabling thin-film efficiencies above 24% with a cost below 20 cents per Watt by 2025, followed by efficiencies above 26% and cost below 15 cents per Watt by 2030.[123]
Durability and lifetime edit
One of the significant drawbacks of thin-film solar cells as compared to mono crystalline modules is their shorter lifetime, though to extent to which this is an issue varies by material with the more established thin-film materials generally having longer lifetimes. The standard lifetime of mono crystalline silicon panels is typically taken to be 30 years[79] with performance degradation rates of around 0.5% per year.[124] Amorphous silicon thin-films tend to have comparable cell lifetimes[79] with slightly higher performance degradation rates around 1% per year.[124] Chalcogenide technologies like CIGS and CIS tend to have similar lifetimes of 20–30 years[34][63] and performance degradation rates just over 1% per year.[124] Emerging technologies tend to have lower lifetimes. Organic photovoltaics had a maximum reported lifetime of 7 years and an average of 5 years in 2016,[67] but typical lifetimes have increased to the range of 15–20 years as of 2020.[125] Similarly, dye-sensitized cells had a maximum reported lifetime of 10 years in 2007,[34] but typical lifetimes have increased to 15–30 years as of 2020.[125] Perovskite cells tend to have short lifetimes, with 5 years being a typical lifetime as of 2016.[68] The lifetime of quantum dot solar cells is unclear due to their developing nature, with some predicting lifetimes to reach 25 years[34] and others setting a realistic lifetime as somewhere between 1 and 10 years.[125]
Some thin-film modules also have issues with degradation under various conditions. Nearly all solar cells experience performance decreases with increasing temperature over a reasonable range of operating temperatures. Established thin-film materials may experience smaller temperature-dependent performance decreases, with amorphous silicon being slightly more resistant than mono crystalline silicon, CIGS more resistant than amorphous silicon, and CdTe displaying the best resistance to performance degradation with temperature.[80] Dye-sensitized solar cells are particularly sensitive to operating temperature, as high temperatures may cause the electrolyte solution to leak and low temperatures may cause it to freeze, leaving the cell inoperable. Perovskite cells also tend to be unstable at high temperatures and may even undergo structural changes that impact the operation of the devices.[64] Beyond temperature-induced degradation, amorphous silicon panels additionally experience light-induced degradation, as do organic photovoltaic cells to an even larger extent.[124][64] Quantum dot cells degrade when exposed to moisture or UV radiation. Similarly, perovskite cells are chemically unstable and degrade when exposed to high temperatures, light, moisture, or UV radiation.[64] Organic cells are also generally considered somewhat unstable,[64] though improvement has been made on the durability organic cells and as of 2022, flexible organic cells have been developed that can be unrolled 500 times without significant performance losses.[126] Unlike other thin-film materials, CdTe tends to be fairly resilient to environmental conditions like temperature and moisture, but flexible CdTe panels may experience performance degradation under applied stresses or strains.[64]
Environmental and health impact edit
In order to meet international renewable energy goals, the worldwide solar capacity must increase significantly. For example, to keep up with the International Energy Agency's goal of 4674 GW of solar capacity installed globally by 2050, significant expansion is required from the 1185 GW installed globally as of 2022.[127] As thin-film solar cells have become more efficient and commercially-viable, it has become clear that they will play an important role in meeting these goals. As such, it's become increasingly important to understand their cumulative environmental impact, both to compare between existing technologies and to identify key areas for improvement in developing technologies. For instance, to evaluate the effect of relatively shorter device lifetimes as compared to established solar modules, and to see whether increasing efficiencies or increasing device lifetimes has a large influence on the total environmental impact of the technologies. Beyond key factors like greenhouse gas (GHG) emissions, questions have been raised about the environmental and health impacts of potentially toxic materials like cadmium that are used in many solar cell technologies. Many scientists and environmentalists have used life cycle analysis as a way to address these questions.[77]
Life cycle analysis edit
Life cycle analysis (LCA) is a family of approaches that attempt to assess the total environmental impact of a product or process in an objective way, from the gathering of raw materials and the manufacturing process all the way to the disposal of the outcome and any waste products. Though LCA approaches aim to be unbiased, the outcome of LCA studies can be sensitive to the particular approach and data used. It is therefore important that LCA findings clearly state the assumptions made any which processes are included and excluded. This will often be specified using the system boundary and life cycle inventory framework. Due to the emerging nature of new photovoltaic technologies, the disposal process may sometimes be left out of a life cycle analysis due to the high uncertainty. In this case, the assessment is referred to as "cradle-to-gate" rather than "cradle-to-grave," because the calculated impact does not cover the full life cycle of the product. However, such studies may miss important environmental impacts from the disposal process, both negative (as in the case of incineration of end-of-life cells and waste products) and positive (as in the case of recycling). It's also important to include the effect of balance of service (BOS) steps, which include transportation, installation, and maintenance as they may also be costly in terms of materials and electricity.[77]
LCA studies can be used to quantify many potential environmental impacts, from land use to transportation-related emissions. Categories of environmental impacts may be grouped into so-called impact factors for standardized quantitative comparison. For solar cells, perhaps the most important impact factor is the total lifetime greenhouse gas (GHG) emission. This is often reported in terms of the global warming potential (GWP), which gives a more direct indication of the environmental impact.[63]
Another important measure of environmental impact is the primary energy demand (PED) which measures the energy (usually electricity) required to produce a particular solar cell. A more useful measure may be the cumulative energy demand (CED), which quantifies the total amount of energy required to produce, use, and dispose of a particular product over its entire lifetime. Relatedly, the energy payback time (EPBT) measures the operational time needed for a solar cell to produce enough energy to account for its cumulative energy demand. Similarly, the carbon payback time (CPBT) measures the operational time needed for a solar cell to produce enough electricity that the avoided carbon emissions from the same amount of electricity generated with the usual energy mix is equal to the amount of carbon emissions the cell will generate over its lifetime. In other words, CPBT measures the time a solar cell needs to run in order to mitigate its own carbon emissions.[67]
These quantities depends on many factors, including where the solar cell is manufactured and deployed, as the typical energy mix varies from place to place. Therefore, the electricity-related emissions from the production process as well as the avoided electricity-related emissions from the solar-generated electricity during operation of the cell can vary depending on the particular module and application. The emissions from a cell may also depend on how the module is deployed, not just because of the raw materials and energy costs associated with the production of mounting hardware, but also from any avoided emissions from replaced building materials, as in the case of building-integrated photovoltaics where solar panels may replace building materials like roof tiles.[79]
Though energy-usage and emissions-related impacts are vital for evaluation of and comparison between technologies, they are not the only important quantities for evaluating the environmental impact of solar cells. Other important impact factors include toxic heavy metal emissions, metal depletion, human toxicity, various eco-toxicities (marine, freshwater, terrestrial), and acidification potential which measures the emission of sulfur and nitrogen oxides.[67][34] Including a wide range of environmental impacts in a life cycle analysis is necessarily to minimize the chance of passing environmental impact from a prominent impact factor like greenhouse gas emission to a less prominent but still relevant impact factor like human toxicity.[77]
Greenhouse gas emissions edit
Using established first-generation mono crystalline silicon solar cells as a benchmark, some thin-film solar cells tend to have lower environmental impacts across most impact factors, however low efficiencies and short lifetimes can increase the environmental impacts of emerging technologies above those of first-generation cells. A standardized measure of greenhouse gas emissions, is displayed in the chart in units of grams of CO2 equivalent emissions per kiloWatt-hour of electricity production for a variety of thin-film materials.[34][79][125] Crystalline silicon is also included for comparison.
In terms of greenhouse gas emissions only, the two most ubiquitous thin-film technologies, amorphous silicon and CdTe, both have significantly lower global warming potential (GWP) than mono crystalline silicon solar cells, with amorphous silicon panels having GWP around 1/3 lower and CdTe nearly 1/2 lower.[68][79] Organic photovoltaics have the smallest GWP of all thin-film PV technologies, with over 60% lower GWP than mono crystalline silicon.[67]
However, this is not the case for all thin-film materials. For many emerging technologies, low efficiencies and short device lifetimes may cause significant increases in environmental impact. Both emerging chalcogenide technologies and established chalcogenide technologies like CIS and CIGS have higher Global warming potential than mono crystalline silicon, as do dye-sensitized and quantum dot solar cells. For antimony-based chalcogenide cells, favorable for their use of less-toxic materials in the manufacturing process, low efficiencies and therefore larger area requirements for solar cells are the driving factor in the increased environmental impact, and cells with modestly improved efficiencies have the potential to outperform mono crystalline silicon in all relevant environmental impact factors. Improving efficiencies for these and other emerging chalcogenide cells is therefore a priority.[63] Low realized efficiencies are also the driving factor behind the relatively large GWP of quantum dot solar cells, despite the potential for these materials to exhibit multiple exciton generation (MEG) from a single photon. Higher efficiencies would also allow for the use of a thinner active layer, reducing both materials costs for the quantum dots themselves and saving on materials and emissions related to encapsulation material. Realizing this potential and thereby increasing efficiency is also a priority for reducing the environmental impact of these cells.[34]
For organic photovoltaics, short lifetimes are instead the driving factor behind GWP. Despite overall impressive performance of OPV relative to other solar technologies, when considering cradle-to-gate rather than cradle-to-grave (i.e. looking only at the material extraction and production processes, discounting the useful lifetime of the solar cells) GWP, OPV constitute a 97% reduction in GHG emissions compared to mono crystalline silicon and 92% reduction relative to amorphous silicon thin-films. This is significantly better than the 60% reduction compared to mono crystalline silicon currently realized, and therefore improving OPV cell lifetimes is a priority for decreasing overall environmental impact.[67] For Perovskite solar cells, with short lifetimes of only around 5 years, this effect may be even more significant. Perovskite solar cells (not included in the chart) typically have significantly larger global warming potential than other thin-film materials in cradle-to-grave LCA, around 5-8x worse than mono crystalline silicon at 150g CO2-eq /kWh. However, in grade-to-gate LCA, Perovskite cells perform 10-30% lower than mono-crystalline silicon, highlighting the importance of the increased environmental impact associated with the need to produce and dispose of multiple Perovskite panels to generate the same amount of electricity as a single mono crystalline silicon panel due to this short lifetime. Increasing the lifetime of Perovskite solar modules is therefore a top priority for decreasing their environmental impact.[68] Other renewable energy sources like wind, nuclear, and hydropower may achieve smaller GWP than some PV technologies.[34]
It's important to note that although emerging thin-film materials don't outperform mono crystalline silicon cells in terms of global warming potential, they still constitute far lower carbon emissions than non-renewable energy sources which have global warming potentials ranging from comparatively clean natural gas with 517g CO2-eq /kWh to the worst polluter lignite with over 1100g CO2-eq /kWh. Thin-film cells also significantly outperform the typical energy mix, which is often in the range of 400-800g CO2-eq /kWh.[79]
| PV Technology | GWP (g CO2-eq/kWh) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| DSPV | 59.8
| ||||||||
| CZGeSe | 53.3
| ||||||||
| CIS | 43.4
| ||||||||
| Sb2Se3 | 40.7
| ||||||||
| Sb2S3 | 40.5
| ||||||||
| CZTS | 36.6
| ||||||||
| CZTSe | 34.9
| ||||||||
| QDPV | 34.6
| ||||||||
| Zn3P2 | 30
| ||||||||
| CIGS | 27.1
| ||||||||
| m-Si* | 25.5
| ||||||||
| a-Si | 15.7
| ||||||||
| CdTe | 14.1
| ||||||||
| OPV | 9.55
| ||||||||
|
*Not thin-film, provided for comparison | |||||||||
The largest contributor to most impact factors, including the global warming potential, is nearly always energy use during the manufacturing process, greatly outweighing other potential sources of environmental impact such as transportation cost and material sourcing.[79] For CIGS cells, for example, this accounts for 98% of the global warming potential, most of which is due to the manufacturing of the absorber layer specifically.[63] In general, for processes that include metal deposition, this is often a particularly significant environmental impact hotspot.[77] For quantum dot photovoltaics, hazardous waste disposal for the solvents used during the manufacturing process also contributes significantly.[34] The level of global warming potential associated with electricity use can vary significantly depending on the location manufacturing takes place, in particular the proportion of renewable to non-renewable energy sources used in the local energy mix.[79]
Energy payback time edit
In general, thin-film panels take less energy to produce than mono crystalline silicon panels,[79] especially as some emerging thin-film technologies have the potential for efficient and cheap roll-to-roll processing.[34] As a result, thin-film technologies tend to fare better than mono crystalline silicon in terms of energy payback time, though amorphous silicon panels are an exception. Thin-film cells typically have lower efficiencies than mono crystalline solar cells, so this effect is largely due to the comparatively lower primary energy demand (PED) associated with producing the cells.
| PV Technology | EPBT (years) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| a-Si | 2.75
| ||||||||
| m-Si | 2.39
| ||||||||
| CIS | 1.88
| ||||||||
| Perovskite | 1.25
| ||||||||
| OPV | 1.21
| ||||||||
| DSPV | 1.19
| ||||||||
| QDPV | 0.99
| ||||||||
| CdTe | 0.738
| ||||||||
|
*Not thin-film, provided for comparison | |||||||||
The application in which the modules are used and the recycling process (if any) for the materials can also play a large role in the overall energy efficiency and greenhouse gas emissions over the lifetime of the cell. Integrating the modules into building design may lead to a large reduction in the environmental impact of the cells due to the avoided emissions related to producing the usual building materials, for example the avoided emissions from roof tile production for a building-integrated solar roof.[79] This effect is especially important for thin-film solar cells, whose lightweight and flexible nature lends itself naturally to building-integrated photovoltaics.[77] 70-90% lower emissions in portable charging applications.[67] This effect holds for some other applications as well, for example organic photovoltaics have 55% lower emissions than crystalline silicon in solar panel applications but up to nearly Similarly, avoided emissions from recycling solar cell components rather than gathering and processing new materials can lead to significantly lower cumulative energy consumption and greenhouse gas emissions. Recycling processes are available for several components of mono crystalline solar cells as well as the glass substrate, CdTe, and CdS in CdTe solar cells.[125] For panels without recycling processes, and particularly for panels with short lifetimes like organic photovoltaics, the disposal of panels may contribute significantly to the environmental impact, and there may be little difference in environmental impact factors if the panel is incinerated or sent to landfill.[67]
Heavy-metal emission and human toxicity edit
Though material selection and extraction does not play a large role in global warming potential, where electricity usage in the manufacturing process is near universally the largest contributor, it often has a significant impact on other important environmental impact factors, including human toxicity, heavy-metal emissions, acidification potential, and metal and ozone depletion.
Human toxicity and heavy-metal emissions are particularly important impact factors for thin-film solar cell production, as the potential environmental and health effect of cadmium use has been a particular concern since the introduction of CdTe cells to the commercial market in the 1990s, when the hazards of cadmium-containing compounds were not well-understood.[128] Public concern over CdTe solar cells has continues as they have become more common.[129] Cadmium is a highly hazardous material[130] that causes kidney, bone, and lung damage and is thought to increase the risk of developing cancer.[131] Initially, all cadmium-containing compounds were classified as hazardous, although we now know that despite both Cd and Te being hazardous separately, the combination CdTe is very chemically stable[25] with a low solubility and presents minimal risk to human health.[128]
Feedstock Cd presents a larger risk, as do pre-cursor materials like CdS, and cadmium acetate, which are frequently used in other photovoltaic cells as well, and often contribute significantly to environmental impact factors such as human toxicity and heavy metal emission.[63] These effects may be more pronounced for nanofabrication processes that produce Cd ions in solution, like the manufacture of quantum dots for QDPV.[34] Due to these effects, CdTe solar cell production is actually seen to have lower heavy-metal emissions than other thin-film solar manufacturing. In fact, CdTe production has lower cadmium emission than ribbon silicon, multi-crystalline silicon, mono-crystalline silicon, or quantum dot PV manufacturing, as well as lower emission of nickel, mercury, arsenic, chromium, and lead.[34] In terms of total heavy metal emissions, quantum dot PV has the highest emissions of PV materials with approximately 0.01 mg/kWh, but still has lower total heavy metal emission than any other renewable or non-renewable electricity source, as shown in the chart.
| Energy source | Total heavy metal emissions (mg/kWh) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Oil | 3.9
| ||||||||
| Wind | 0.61
| ||||||||
| Diesel | 0.32
| ||||||||
| Coal | 0.24
| ||||||||
| Lignite | 0.22
| ||||||||
| Nuclear | 0.13
| ||||||||
| Hydro | 0.039
| ||||||||
| Natural gas | 0.029
| ||||||||
| QDPV | 0.010
| ||||||||
|
Data from [34] | |||||||||
The desire to alleviate safety concerns around cadmium and CdTe solar cells specifically has sparked the development of other chalcogenide PV materials that are non-toxic or less toxic, particularly antimony-based chalcogenides. In these emerging chalcogenide cells, the use of CdS is the largest contribution to impact factors like human toxicity and metal depletion, though stainless steel also contributes significantly to the impact of these and other PV materials. In CIGS cells, for example, stainless steel accounts for 80% of the total toxicity associated with cell production and also contributes significantly to ozone depletion.[63]
| Energy source | HT (kg 1.4DBeq/kWh) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Lignite | 1.9
| ||||||||
| Coal | 0.24
| ||||||||
| CZGeSe | 0.018
| ||||||||
| Perovskite | 0.018
| ||||||||
| Sb2Se3 | 0.018
| ||||||||
| Sb2S3 | 0.018
| ||||||||
| CZTS | 0.013
| ||||||||
| CZTSe | 0.012
| ||||||||
| m-Si | 0.0069
| ||||||||
| OPV-D | 0.0021
| ||||||||
Another potential impact factor of interest for PV manufacturing is the acidification potential, which quantifies the emission of sulfur and nitrogen oxides which contribute to the acidification of soil, freshwater, and the ocean and their negative environmental effects. In this respect, QDPV has the lowest emissions, with CdTe being a close second.[34]
See also edit
References edit
- ^ a b "Best Research-Cell Efficiency Chart". www.nrel.gov. Retrieved April 5, 2023.
- ^ a b "PHOTOVOLTAICS REPORT" (PDF). Fraunhofer Institute for Solar Energy Systems, ISE.
- ^ a b Feldman, David; Dummit, Krysta; Jarett, Zuboy; Margolis, Robert (July 12, 2022). "Summer 2022 Solar Industry Update" (PDF). National Renewable Energy Laboratory.
- ^ Madou, Marc J. (2011). From MEMS to Bio-MEMS and Bio-NEMS: Manufacturing Techniques and Applications. CRC Press. p. 246. ISBN 9781439895245.
- ^ "The Nobel Prize in Physics 2000". NobelPrize.org. Retrieved March 27, 2023.
- ^ "History". Institute of Energy Conversion. Archived from the original on March 27, 2023. Retrieved March 26, 2023.
- ^ "Karl Wolfgang Böer papers | Manuscript and Archival Collection Finding Aids". library.udel.edu. Retrieved March 27, 2023.
- ^ Landis, Geoffrey A.; Bailey, Sheila G.; Flood, Dennis J. (June 5, 1989). "Advances in Thin-Film Solar Cells for Lightweight Space Photovoltaic Power" (PDF). NASA Technical Reports Server (NTRS). National Aeronautics and Space Administration (NASA), Lewis Research Center.
- ^ Wallace, W. L.; Sabisky, E. S. (August 1986). "The Status and Future of Government-Supported Amorphous Silicon Research in the United States" (PDF). National Renewable Energy Laboratory (NREL).
- ^ "The History of Solar" (PDF). U.S. Department of Energy Office of Energy Efficiency and Renewable Energy. Archived (PDF) from the original on September 11, 2021. Retrieved March 26, 2023.
- ^ Watts, R. L.; Smith, S. A.; Dirks, J. A. (April 1985). "Photovoltaic Industry Progress through 1984" (PDF). U.S. Department of Energy Office of Scientific and Technical Information. doi:10.2172/5685828. Retrieved March 26, 2023.
{{cite journal}}: Unknown parameter|agency=ignored (help) - ^ a b c "The History of Solar" (PDF). U.S. Department of Energy Office of Energy Efficiency and Renewable Energy. Archived (PDF) from the original on September 11, 2021. Retrieved March 26, 2023.
- ^ "World's Most Efficient Solar Cell". www.nrel.gov. Retrieved March 27, 2023.
- ^ "4 times square building - integrated photovoltaic system" (PDF). Kiss + Cathcart, Architects. Retrieved March 26, 2023.
- ^ Bauhuis, G. J.; Mulder, P.; Schermer, J. J.; Haverkamp, E. J.; Deelen, J. van; Larsen, P. K. (2005). "HIGH EFFICIENCY THIN FILM GaAs SOLAR CELLS WITH IMPROVED RADIATION HARDNESS". 20th European Photovoltaic Solar Energy Conference: 468–471.
- ^ a b Kamat, Prashant V. (October 18, 2008). "Quantum Dot Solar Cells. Semiconductor Nanocrystals as Light Harvesters". Journal of Physical Chemistry C. 112 (48): 18737–18753. doi:10.1021/jp806791s.
- ^ O'Regan, Brian; Grätzel, Michael (October 1991). "A low-cost, high-efficiency solar cell based on dye-sensitized colloidal TiO2 films". Nature. 353 (6346): 737–740. Bibcode:1991Natur.353..737O. doi:10.1038/353737a0. ISSN 1476-4687. S2CID 4340159.
- ^ "Quantum Dots Promise to Significantly Boost Solar Cell Efficiencies" (PDF). National Renewable Energy Laboratory (NREL). August 2013.
- ^ Kojima, Akihiro; Teshima, Kenjiro; Shirai, Yasuo; Miyasaka, Tsutomu (May 6, 2009). "Organometal halide perovskites as visible-light sensitizers for photovoltaic cells". Journal of the American Chemical Society. 131 (17): 6050–6051. doi:10.1021/ja809598r. ISSN 1520-5126. PMID 19366264.
- ^ Editorial, BCC Research. "A History of Perovskite Solar Cells". blog.bccresearch.com. Retrieved March 28, 2023.
- ^ Lee, Brendon (June 10, 2015). "Printed solar cells hold promise for unlit rural areas - Asia & Pacific". SciDevNet. Archived from the original on April 1, 2022.
- ^ "Solar cells as light as a soap bubble". MIT News | Massachusetts Institute of Technology. February 26, 2016. Retrieved March 27, 2023.
- ^ a b "Paper-thin solar cell can turn any surface into a power source". MIT News | Massachusetts Institute of Technology. December 9, 2022. Retrieved March 27, 2023.
- ^ Saravanapavanantham, Mayuran; Mwaura, Jeremiah; Bulović, Vladimir (December 9, 2022). "Printed Organic Photovoltaic Modules on Transferable Ultra-thin Substrates as Additive Power Sources". Small Methods. 7 (1): 2200940. doi:10.1002/smtd.202200940. ISSN 2366-9608. PMID 36482828. S2CID 254524625.
- ^ a b c "PHOTOVOLTAICS REPORT" (PDF). Fraunhofer Institute for Solar Energy Systems, ISE.
- ^ "photovoltaics-report-slides.pdf" (PDF). FRAUNHOFER INSTITUTE FOR SOLAR ENERGY SYSTEMS ISE. November 7, 2013. Archived from the original (PDF) on July 25, 2014.
- ^ Feldman, David; Dummit, Krysta; Jarett, Zuboy; Margolis, Robert (July 12, 2022). "Summer 2022 Solar Industry Update" (PDF). National Renewable Energy Laboratory.
- ^ "PV Cells 101, Part 2: Solar Photovoltaic Cell Research Directions". Energy.gov. Retrieved April 5, 2023.
- ^ "The photovoltaic effect | PVEducation". www.pveducation.org. Retrieved April 5, 2023.
- ^ Kangsabanik, Jiban; Svendsen, Mark Kamper; Taghizadeh, Alireza; Crovetto, Andrea; Thygesen, Kristian S. (November 2, 2022). "Indirect Band Gap Semiconductors for Thin-Film Photovoltaics: High-Throughput Calculation of Phonon-Assisted Absorption". Journal of the American Chemical Society. 144 (43): 19872–19883. arXiv:2207.09569. doi:10.1021/jacs.2c07567. ISSN 0002-7863. PMID 36270007. S2CID 250699345.
- ^ Pearce, J.; Lau, A. (2002). "Net Energy Analysis for Sustainable Energy Production from Silicon Based Solar Cells" (PDF). Solar Energy. pp. 181–186. doi:10.1115/SED2002-1051. ISBN 978-0-7918-1689-9.
- ^ Datasheets of the market leaders: First Solar for thin film, Suntech and SunPower for crystalline silicon
- ^ a b c "Photovoltaics Report" (PDF). Fraunhofer ISE. July 28, 2014. Archived (PDF) from the original on August 9, 2014. Retrieved August 31, 2014.
- ^ a b c d e f g h i j k l m n o p q r Şengül, Hatice; Theis, Thomas L. (January 1, 2011). "An environmental impact assessment of quantum dot photovoltaics (QDPV) from raw material acquisition through use". Journal of Cleaner Production. 19 (1): 21–31. doi:10.1016/j.jclepro.2010.08.010. ISSN 0959-6526.
- ^ First Solar Seriers 6 Datasheet, March 5, 2021
- ^ Fthenakis, Vasilis M. (2004). "Life cycle impact analysis of cadmium in CdTe PV production" (PDF). Renewable and Sustainable Energy Reviews. 8 (4): 303–334. doi:10.1016/j.rser.2003.12.001. Archived (PDF) from the original on May 8, 2014.
- ^ Werner, Jürgen H. (November 2, 2011). "TOXIC SUBSTANCES IN PHOTOVOLTAIC MODULES" (PDF). postfreemarket.net. Institute of Photovoltaics, University of Stuttgart, Germany – The 21st International Photovoltaic Science and Engineering Conference 2011 Fukuoka, Japan. p. 2. Archived (PDF) from the original on December 21, 2014. Retrieved September 23, 2014.
- ^ "The Lowdown on the Safety of First Solar's CdTe Thin Film". www.greentechmedia.com.
- ^ "Cadmium: The Dark Side of Thin-Film? – Old GigaOm". old.gigaom.com.
- ^ "Supply Constraints Analysis, National Renewable Energy Laboratory".
- ^ Green, M. A.; et al. (2023). "Solar cell efficiency tables (version 62)". Progress in Photovoltaics. 31 (7): 651–663. doi:10.1002/pip.3726.
- ^ "Press release" (PDF). AVANCIS GmbH.
- ^ "Flexible Solarzellen mit Rekordwirkungsgrad von 22,2%". Empa.
- ^ "Large Area Flexible PV Modules Show Equivalent c-Si Performance". MiaSolé.
- ^ "Photovoltaics report" (PDF). Fraunhofer Institute for Solar Energy Systems (ISE).
- ^ Witte, W.; et al. (2014). "Substitution of the CdS buffer layer in CIGS thin-film solar cells". Vakuum in Forschung und Praxis. 26: 23–27. doi:10.1002/vipr.201400546.
- ^ Shin, M. J.; et al. (2021). "Bifacial photovoltaic performance of semitransparent ultrathin Cu(In,Ga)Se2 solar cells with front and rear transparent conducting oxide contacts". Applied Surface Science. 535: 147732. Bibcode:2021ApSS..53547732S. doi:10.1016/j.apsusc.2020.147732.
- ^ a b "CIGS White Paper" (PDF). Zentrum für Sonnenenergie- und Wasserstoff-Forschung Baden-Württemberg (ZSW).
- ^ a b Green, M. A. (2003), "Crystalline and thin-film silicon solar cells: state of the art and future potential", Solar Energy, 74 (3): 181–192, Bibcode:2003SoEn...74..181G, doi:10.1016/S0038-092X(03)00187-7.
- ^ Photovoltaics. Engineering.Com (July 9, 2007). Retrieved on January 19, 2011.
- ^ "Amorphes Silizium für Solarzellen" (PDF) (in German).
- ^ Arvind Shah et al. (2003): Microcrystalline silicon and micromorph tandem solar cells. In: Solar Energy Materials and Solar Cells, 78, pp. 469-491
- ^ "New Record-Breaking PV Module Efficiency has been achieved". TEL Solar Website. TEL Solar. Retrieved July 14, 2014.
- ^ J. M. Pearce; N. Podraza; R. W. Collins; M.M. Al-Jassim; K.M. Jones; J. Deng & C. R. Wronski (2007). "Optimization of Open-Circuit Voltage in Amorphous Silicon Solar Cells with Mixed Phase (Amorphous + Nanocrystalline) p-Type Contacts of Low Nanocrystalline Content" (PDF). Journal of Applied Physics. 101 (11): 114301–114301–7. Bibcode:2007JAP...101k4301P. doi:10.1063/1.2714507. Archived from the original (PDF) on June 14, 2011. Retrieved June 12, 2009.
- ^ Pearce, J. M.; Podraza, N.; Collins, R. W.; Al-Jassim, M. M.; Jones, K. M.; Deng, J.; Wronski, C. R. (2007). "Optimization of open circuit voltage in amorphous silicon solar cells with mixed-phase (amorphous+nanocrystalline) p-type contacts of low nanocrystalline content" (PDF). Journal of Applied Physics. 101 (11): 114301–114301–7. Bibcode:2007JAP...101k4301P. doi:10.1063/1.2714507. Archived from the original (PDF) on June 14, 2011. Retrieved June 12, 2009.
- ^ "GaAs Solar Cells". sinovoltaics.com. Retrieved November 18, 2020.
- ^ Green, Martin A.; Hishikawa, Yoshihiro; Dunlop, Ewan D.; Levi, Dean H.; Hohl-Ebinger, Jochen; Yoshita, Masahiro; Ho-Baillie, Anita W. Y. (2019). "Solar cell efficiency tables (Version 53)". Progress in Photovoltaics: Research and Applications. 27 (1): 3–12. doi:10.1002/pip.3102. ISSN 1099-159X. S2CID 104439991.
- ^ Nayak, Pabitra K.; Mahesh, Suhas; Snaith, Henry J.; Cahen, David (2019). "Photovoltaic solar cell technologies: analysing the state of the art". Nature Reviews Materials. 4 (4): 269–285. Bibcode:2019NatRM...4..269N. doi:10.1038/s41578-019-0097-0. ISSN 2058-8437. S2CID 141233525.
- ^ a b Massiot, Inès; Cattoni, Andrea; Collin, Stéphane (November 2, 2020). "Progress and prospects for ultrathin solar cells" (PDF). Nature Energy. 5 (12): 959–972. Bibcode:2020NatEn...5..959M. doi:10.1038/s41560-020-00714-4. ISSN 2058-7546. S2CID 228814810.
- ^ Konagai, Makoto; Sugimoto, Mitsunori; Takahashi, Kiyoshi (December 1, 1978). "High efficiency GaAs thin film solar cells by peeled film technology". Journal of Crystal Growth. 45: 277–280. Bibcode:1978JCrGr..45..277K. doi:10.1016/0022-0248(78)90449-9. ISSN 0022-0248.
- ^ Cheng, Cheng-Wei; Shiu, Kuen-Ting; Li, Ning; Han, Shu-Jen; Shi, Leathen; Sadana, Devendra K. (March 12, 2013). "Epitaxial lift-off process for gallium arsenide substrate reuse and flexible electronics". Nature Communications. 4 (1): 1577. Bibcode:2013NatCo...4.1577C. doi:10.1038/ncomms2583. ISSN 2041-1723. PMID 23481385.
- ^ a b c d Efaz, Erteza Tawsif; Rhaman, Md Meganur; Imam, Safayat Al; Bashar, Khandaker Lubaba; Kabir, Fahmid; Mourtaza, MD Ehasan; Sakib, Syed Nazmus; Mozahid, F. A. (September 23, 2021). "A review of primary technologies of thin-film solar cells". Engineering Research Express. 3 (3): 032001. Bibcode:2021ERExp...3c2001E. doi:10.1088/2631-8695/ac2353. ISSN 2631-8695. S2CID 237613178.
- ^ a b c d e f g h i j Resalati, Shahaboddin; Okoroafor, Tobechi; Maalouf, Amani; Saucedo, Edgardo; Placidi, Marcel (May 1, 2022). "Life cycle assessment of different chalcogenide thin-film solar cells". Applied Energy. 313: 118888. Bibcode:2022ApEn..31318888R. doi:10.1016/j.apenergy.2022.118888. hdl:2117/370823. ISSN 0306-2619. S2CID 247433824.
- ^ a b c d e f g h i j Sivaraj, Santhosh; Rathanasamy, Rajasekar; Kaliyannan, Gobinath Velu; Panchal, Hitesh; Jawad Alrubaie, Ali; Musa Jaber, Mustafa; Said, Zafar; Memon, Saim (November 19, 2022). "A Comprehensive Review on Current Performance, Challenges and Progress in Thin-Film Solar Cells" (PDF). Energies. 15 (22): 8688. doi:10.3390/en15228688. ISSN 1996-1073.
- ^ Grätzel, Michael (October 31, 2003). "Dye-sensitized solar cells". Journal of Photochemistry and Photobiology C: Photochemistry Reviews. 4 (2): 145–153. CiteSeerX 10.1.1.687.8125. doi:10.1016/S1389-5567(03)00026-1. ISSN 1389-5567.
- ^ a b c d "Best Research-Cell Efficiency Chart". www.nrel.gov. Retrieved April 6, 2023.
- ^ a b c d e f g h i j k Tsang, Michael P.; Sonnemann, Guido W.; Bassani, Dario M. (November 1, 2016). "Life-cycle assessment of cradle-to-grave opportunities and environmental impacts of organic photovoltaic solar panels compared to conventional technologies". Solar Energy Materials and Solar Cells. Life cycle, environmental, ecology and impact analysis of solar technology. 156: 37–48. doi:10.1016/j.solmat.2016.04.024. ISSN 0927-0248.
- ^ a b c d e f g h Celik, Ilke; Song, Zhaoning; Cimaroli, Alexander J.; Yan, Yanfa; Heben, Michael J.; Apul, Defne (November 1, 2016). "Life Cycle Assessment (LCA) of perovskite PV cells projected from lab to fab". Solar Energy Materials and Solar Cells. Life cycle, environmental, ecology and impact analysis of solar technology. 156: 157–169. doi:10.1016/j.solmat.2016.04.037. ISSN 0927-0248.
- ^ "A new stable and cost-cutting type of perovskite solar cell". PHYS.org. July 17, 2014. Retrieved August 4, 2015.
- ^ "Spray-deposition steers perovskite solar cells towards commercialisation". ChemistryWorld. July 29, 2014. Retrieved August 4, 2015.
- ^ "Perovskite Solar Cells". Ossila. Retrieved August 4, 2015.
- ^ Huang, Xinjing; Fan, Dejiu; Li, Yongxi; Forrest, Stephen R. (July 20, 2022). "Multilevel peel-off patterning of a prototype semitransparent organic photovoltaic module". Joule. 6 (7): 1581–1589. doi:10.1016/j.joule.2022.06.015. ISSN 2542-4785. OSTI 1878096. S2CID 250541919.
- Lay summary: "Toward manufacturing semitransparent solar cells the size of windows". University of Michigan. Retrieved August 31, 2022.
- ^ "Transparent solar panels for windows hit record 8% efficiency". University of Michigan News. August 17, 2020. Retrieved August 23, 2022.
- ^ Li, Yongxi; Guo, Xia; Peng, Zhengxing; Qu, Boning; Yan, Hongping; Ade, Harald; Zhang, Maojie; Forrest, Stephen R. (September 2020). "Color-neutral, semitransparent organic photovoltaics for power window applications". Proceedings of the National Academy of Sciences. 117 (35): 21147–21154. Bibcode:2020PNAS..11721147L. doi:10.1073/pnas.2007799117. ISSN 0027-8424. PMC 7474591. PMID 32817532.
- ^ "Researchers fabricate highly transparent solar cell with 2D atomic sheet". Tohoku University. Retrieved August 23, 2022.
- ^ He, Xing; Iwamoto, Yuta; Kaneko, Toshiro; Kato, Toshiaki (July 4, 2022). "Fabrication of near-invisible solar cell with monolayer WS2". Scientific Reports. 12 (1): 11315. Bibcode:2022NatSR..1211315H. doi:10.1038/s41598-022-15352-x. ISSN 2045-2322. PMC 9253307. PMID 35787666.
- ^ a b c d e f Chatzisideris, Marios D.; Espinosa, Nieves; Laurent, Alexis; Krebs, Frederik C. (November 1, 2016). "Ecodesign perspectives of thin-film photovoltaic technologies: A review of life cycle assessment studies" (PDF). Solar Energy Materials and Solar Cells. Life cycle, environmental, ecology and impact analysis of solar technology. 156: 2–10. doi:10.1016/j.solmat.2016.05.048. ISSN 0927-0248. S2CID 99033877.
- ^ "4 times square building - integrated photovoltaic system" (PDF). Kiss + Cathcart, Architects. Retrieved March 26, 2023.
- ^ a b c d e f g h i j k l m Kittner, Noah; Gheewala, Shabbir H.; Kamens, Richard M. (December 1, 2013). "An environmental life cycle comparison of single-crystalline and amorphous-silicon thin-film photovoltaic systems in Thailand". Energy for Sustainable Development. 17 (6): 605–614. Bibcode:2013ESusD..17..605K. doi:10.1016/j.esd.2013.09.003. ISSN 0973-0826.
- ^ a b Efaz, Erteza Tawsif; Rhaman, Md Meganur; Imam, Safayat Al; Bashar, Khandaker Lubaba; Kabir, Fahmid; Mourtaza, MD Ehasan; Sakib, Syed Nazmus; Mozahid, F. A. (September 1, 2021). "A review of primary technologies of thin-film solar cells". Engineering Research Express. 3 (3): 032001. Bibcode:2021ERExp...3c2001E. doi:10.1088/2631-8695/ac2353. ISSN 2631-8695. S2CID 237613178.
- ^ Casey, Tina (February 27, 2014). "New Thin Film Solar Cell Efficiency Record for First Solar". CleanTechnica.
- ^ "Panasonic HIT Solar Cell Sets World Efficiency Record −". April 19, 2014.
- ^ "World Record Thin-Film Solar Cell Efficiency of 22.3% achieved by solar frontier – Renew India Campaign – solar photovoltaic, Indian Solar News, Indian Wind News, Indian Wind Market". www.renewindians.com. Retrieved December 14, 2015.
- ^ Lindholm; Fossum, J. G.; Burgess, E. L. (1979). "Application of the superposition principle to solar-cell analysis". IEEE Transactions on Electron Devices. 26 (3): 165–171. doi:10.1109/T-ED.1979.19400. ISSN 0018-9383. S2CID 25916176.
- ^ "Quantum Efficiency | PVEducation". www.pveducation.org. Retrieved April 5, 2023.
- ^ Xu, Yunlu; Gong, Tao; Munday, Jeremy N. (September 2, 2015). "The generalized Shockley-Queisser limit for nanostructured solar cells". Scientific Reports. 5 (1): 13536. arXiv:1412.1136. Bibcode:2015NatSR...513536X. doi:10.1038/srep13536. ISSN 2045-2322. PMC 4557037. PMID 26329479. S2CID 17029239.
- ^ Cite error: The named reference
:14was invoked but never defined (see the help page). - ^ National Renewable Energy Laboratory (NREL) (February 2005). "PV FAQs: What's New in Concentrating PV?" (PDF). National Renewable Energy Laboratory (NREL).
- ^ Widenborg, Per I.; Aberle, Armin G. (2007). "Polycrystalline Silicon Thin-Film Solar Cells on AIT-Textured Glass Superstrates" (PDF). Advances in OptoElectronics. 2007: 1–7. doi:10.1155/2007/24584.
- ^ "Renewable Sources". www.customwritingtips.com. Archived from the original on February 23, 2013.
- ^ Xu, Zhida; Yao, Yuan; Brueckner, Eric; Li, Lanfang; Jiang, Jing; Nuzzo, Ralph G.; Liu, Logan (2014). "Black silicon solar thin-film microcells integrating top nanocone structures for broadband and omnidirectional light-trapping". Nanotechnology. 25 (30): 305301. arXiv:1406.1729. Bibcode:2014Nanot..25D5301X. doi:10.1088/0957-4484/25/30/305301. PMID 25006119. S2CID 206069610.
- ^ Mendes, Manuel J.; Sanchez-Sobrado, Olalla; Haque, Sirazul; Mateus, Tiago; Águas, Hugo; Fortunato, Elvira; Martins, Rodrigo (January 1, 2020), Enrichi, Francesco; Righini, Giancarlo C. (eds.), "Chapter Nine - Wave-optical front structures on silicon and perovskite thin-film solar cells", Solar Cells and Light Management, Elsevier, pp. 315–354, ISBN 978-0-08-102762-2, retrieved March 10, 2023
- ^ Schuster, Christian Stefano; Crupi, Isodiana; Halme, Janne; Koç, Mehmet; Mendes, Manuel João; Peters, Ian Marius; Yerci, Selçuk (2022), Lackner, Maximilian; Sajjadi, Baharak; Chen, Wei-Yin (eds.), "Empowering Photovoltaics with Smart Light Management Technologies", Handbook of Climate Change Mitigation and Adaptation, Cham: Springer International Publishing, pp. 1165–1248, doi:10.1007/978-3-030-72579-2_112, ISBN 978-3-030-72579-2, retrieved March 10, 2023
- ^ Wu, Jiang; Yu, Peng; Susha, Andrei S.; Sablon, Kimberly A.; Chen, Haiyuan; Zhou, Zhihua; Li, Handong; Ji, Haining; Niu, Xiaobin (April 1, 2015). "Broadband efficiency enhancement in quantum dot solar cells coupled with multispiked plasmonic nanostars". Nano Energy. 13: 827–835. doi:10.1016/j.nanoen.2015.02.012. S2CID 98282021.
- ^ Yu, Peng; Yao, Yisen; Wu, Jiang; Niu, Xiaobin; Rogach, Andrey L.; Wang, Zhiming (August 9, 2017). "Effects of Plasmonic Metal Core -Dielectric Shell Nanoparticles on the Broadband Light Absorption Enhancement in Thin Film Solar Cells". Scientific Reports. 7 (1): 7696. Bibcode:2017NatSR...7.7696Y. doi:10.1038/s41598-017-08077-9. ISSN 2045-2322. PMC 5550503. PMID 28794487.
- ^ Morawiec, Seweryn; Mendes, Manuel J.; Filonovich, Sergej A.; Mateus, Tiago; Mirabella, Salvatore; Águas, Hugo; Ferreira, Isabel; Simone, Francesca; Fortunato, Elvira; Martins, Rodrigo; Priolo, Francesco; Crupi, Isodiana (June 30, 2014). "Broadband photocurrent enhancement in a-Si:H solar cells with plasmonic back reflectors". Optics Express. 22 (S4): A1059-70. Bibcode:2014OExpr..22A1059M. doi:10.1364/OE.22.0A1059. hdl:10362/35672. ISSN 1094-4087. PMID 24978069.
- ^ Mendes, Manuel J.; Morawiec, Seweryn; Mateus, Tiago; Lyubchyk, Andriy; Águas, Hugo; Ferreira, Isabel; Fortunato, Elvira; Martins, Rodrigo; Priolo, Francesco; Crupi, Isodiana (March 11, 2015). "Broadband light trapping in thin film solar cells with self-organized plasmonic nano-colloids". Nanotechnology. 26 (13): 135202. Bibcode:2015Nanot..26m5202M. doi:10.1088/0957-4484/26/13/135202. ISSN 0957-4484. PMID 25760231. S2CID 24473789.
- ^ Terry, Mason L.; Straub, Axel; Inns, Daniel; Song, Dengyuan; Aberle, Armin G. (2005). "Large open-circuit voltage improvement by rapid thermal annealing of evaporated solid-phase-crystallized thin-film silicon solar cells on glass". Applied Physics Letters. 86 (17): 172108. Bibcode:2005ApPhL..86q2108T. doi:10.1063/1.1921352.
- ^ Yan, Baojie; Yue, Guozhen; Sivec, Laura; Yang, Jeffrey; Guha, Subhendu; Jiang, Chun-Sheng (2011). "Innovative dual function nc-SiOx:H layer leading to a >16% efficient multi-junction thin-film silicon solar cell". Applied Physics Letters. 99 (11): 11351. Bibcode:2011ApPhL..99k3512Y. doi:10.1063/1.3638068.
- ^ Yu, Peng; Wu, Jiang; Liu, Shenting; Xiong, Jie; Jagadish, Chennupati; Wang, Zhiming M. (December 1, 2016). "Design and fabrication of silicon nanowires towards efficient solar cells" (PDF). Nano Today. 11 (6): 704–737. doi:10.1016/j.nantod.2016.10.001.
- ^ RenewableEnergyWorld.com How thin film solar fares vs crystalline silicon, 3 January 2011
- ^ Diane Cardwell; Keith Bradsher (January 9, 2013). "Chinese Firm Buys U.S. Solar Start-Up". The New York Times. Retrieved January 10, 2013.
- ^ Andorka, Frank (January 8, 2014). "CIGS Solar Cells, Simplified". solarpowerworldonline.com/. Solar Power World. Archived from the original on August 19, 2014. Retrieved August 16, 2014.
- ^ "South Korean enterprises terminate or downsize thin-film business". OfWeek.com/. July 17, 2014. Archived from the original on May 29, 2015. Retrieved May 29, 2015.
- ^ "Annual Report 2014". IEA-PVPS. May 21, 2015. pp. 49, 78.
Samsung SDI decided to stop the production of CIGS thin film PV modules. Hanergy: Table 3 on page 49
- ^ solar-frontier.com – PressRelease Solar Frontier to build 150 MW 150 MW CIS solar module plant in Tohoku, Japan, December 19, 2013
- ^ "Australia's biggest solar farm opens at Nyngan, western NSW". ABC.net.au. April 18, 2015.
- ^ Peralta, Eyder. (April 7, 2011) GE Unveils Plans To Build Largest Solar Panel Factory In U.S. : The Two-Way. NPR. Retrieved on 2011-05-05.
- ^ PVTECH.org First Solar buys GE’s CdTe thin-film IP and forms business partnership, August 6, 2013
- ^ Raabe, Steve; Jaffe, Mark (November 4, 2012). "Bankrupt Abound Solar of Colo. lives on as political football". Denver Post.
- ^ "The End Arrives for ECD Solar". GreentechMedia. February 14, 2012.
- ^ "Oerlikon Divests Its Solar Business and the Fate of Amorphous Silicon PV". GrrentechMedia. March 2, 2012.
- ^ "Rest in Peace: The List of Deceased Solar Companies". GreenTechMedia. April 6, 2013. Retrieved July 30, 2015.
- ^ "NovaSolar, Formerly OptiSolar, Leaving Smoking Crater in Fremont". GreenTechMedia. February 24, 2012. Retrieved July 30, 2015.
- ^ "Chinese Subsidiary of Suntech Power Declares Bankruptcy". New York Times. March 20, 2013.
- ^ "Suntech Seeks New Cash After China Bankruptcy, Liquidator Says". Bloomberg. April 29, 2014.
- ^ "PVX spot market price index solar PV modules". SolarServer. June 20, 2014. Archived from the original on August 29, 2014. Retrieved July 30, 2015.
- ^ "0.38 EUR to USD - Euros to US Dollars Exchange Rate". www.xe.com.
- ^ "Thin-Film Solar Panels | American Solar Energy Society".
- ^ "Pros and Cons of Metal Roofs for Your Home".
- ^ a b "Solar Panels vs. Thin-Film Laminates: Costs, Pros & Cons, Top Brands". January 19, 2022.
- ^ Zweibel, Ken (September 1995). "Thin Films: Past, Present, Future". Progress in Photovoltaics: Research and Applications. 3 (5): 279–293. doi:10.1002/pip.4670030503.
- ^ "Cadmium Telluride Accelerator Consortium". www.nrel.gov. Retrieved April 6, 2023.
- ^ a b c d Jordan, Dirk C.; Kurtz, Sarah R. (June 2012). "Photovoltaic Degradation Rates -- An Analytical Review" (PDF). National Renewable Energy Laboratory (NREL).
- ^ a b c d e f g Muteri, Vincenzo; Cellura, Maurizio; Curto, Domenico; Franzitta, Vincenzo; Longo, Sonia; Mistretta, Marina; Parisi, Maria Laura (January 2020). "Review on Life Cycle Assessment of Solar Photovoltaic Panels". Energies. 13 (1): 252. doi:10.3390/en13010252. hdl:10447/392647. ISSN 1996-1073.
- ^ "Paper-thin solar cell can turn any surface into a power source". MIT News | Massachusetts Institute of Technology. December 9, 2022. Retrieved April 5, 2023.
- ^ "Solar - 10 Predictions for 2022". BloombergNEF. January 26, 2022. Retrieved April 4, 2023.
- ^ a b Babayigit, Aslihan; Ethirajan, Anitha; Muller, Marc; Conings, Bert (February 24, 2016). "Toxicity of organometal halide perovskite solar cells". Nature Materials. 15 (3): 247–251. Bibcode:2016NatMa..15..247B. doi:10.1038/nmat4572. hdl:2268/195044. ISSN 1476-4660. PMID 26906955.
- ^ "The Lowdown on the Safety of First Solar's CdTe Thin Film". www.greentechmedia.com. Retrieved April 5, 2023.
- ^ Fthenakis, V. M.; Moskowitz, P. D. (March 21, 1995). "Thin-Film Photovoltaic Cells: Health and Environmental Issues in their Manufacture, Use, and Disposal". Progress in Photovoltaics: Research and Applications. 3 (5): 295–306. doi:10.1002/pip.4670030504.
- ^ MOSKOWITZ, PAUL D. (February 23, 2007). "ENVIRONMENTAL, HEALTH AND SAFETY ISSUES RELATED TO THE PRODUCTION AND USE OF CdTe PHOTOVOLTAIC MODULES". International Journal of Solar Energy. 12 (1–4): 259–281. doi:10.1080/01425919208909767. ISSN 1992-1993.
- ^ Burchart-Korol, Dorota; Pustejovska, Pavlina; Blaut, Agata; Jursova, Simona; Korol, Jerzy (November 1, 2018). "Comparative life cycle assessment of current and future electricity generation systems in the Czech Republic and Poland". The International Journal of Life Cycle Assessment. 23 (11): 2165–2177. Bibcode:2018IJLCA..23.2165B. doi:10.1007/s11367-018-1450-z. hdl:10084/132821. ISSN 1614-7502. S2CID 116189232.
Sources edit
- Grama, S. "A Survey of Thin-Film Solar Photovoltaic Industry & Technologies." Massachusetts Institute of Technology, 2008.
- Green, Martin A. "Consolidation of thin-film photovoltaic technology: the coming decade of opportunity.” Progress in Photovoltaics: Research and Applications 14, no. 5 (2006): 383–392.
- Green, M. A. “Recent developments in photovoltaics.” Solar Energy 76, no. 1-3 (2004): 3–8.
- Beaucarne, Guy. “Silicon Thin-Film Solar Cells.” Advances in OptoElectronics 2007 (August 2007): 12.
- Ullal, H. S., and B. von Roedern. “Thin Film CIGS and CdTe Photovoltaic Technologies: Commercialization, Critical Issues, and Applications; Preprint” (2007).
- Hegedus, S. “Thin film solar modules: the low cost, high throughput and versatile alternative to Si wafers.” Progress in Photovoltaics: Research and Applications 14, no. 5 (2006): 393–411.
- Poortmans, J., and V. Arkhipov. Thin Film Solar Cells: Fabrication, Characterization and Applications. Wiley, 2006.
- Wronski, C.R., B. Von Roedern, and A. Kolodziej. “Thin-film Si:H-based solar cells.” Vacuum 82, no. 10 (June 3, 2008): 1145–1150.
- Chopra, K. L., P. D. Paulson, and V. Dutta. “Thin-film solar cells: an overview.” Progress in Photovoltaics: Research and Applications 12, no. 2-3 (2004): 69–92.
- Hamakawa, Y. Thin-Film Solar Cells: Next Generation Photovoltaics and Its Applications. Springer, 2004.
- Green, Martin. “Thin-film solar cells: review of materials, technologies and commercial status.” Journal of Materials Science: Materials in Electronics 18 (October 1, 2007): 15–19.
External links edit
- Solar Panels, Link Solar
- Flexcellence Archived May 10, 2009, at the Wayback Machine, a STReP financed by the Sixth Framework Programme (FP6) of the EU. Full title : Roll-to-roll technology for the production of high-efficiency low cost thin-film silicon photovoltaic modules.