


Summary
In organic chemistry, umpolung (German: [ˈʔʊmˌpoːlʊŋ]) or polarity inversion is the chemical modification of a functional group with the aim of the reversal of polarity of that group.[1][2] This modification allows secondary reactions of this functional group that would otherwise not be possible.[3] The concept was introduced by D. Seebach (hence the German word umpolung for reversed polarity) and E.J. Corey. Polarity analysis during retrosynthetic analysis tells a chemist when umpolung tactics are required to synthesize a target molecule.
Introduction edit
The vast majority of important organic molecules contain heteroatoms, which polarize carbon skeletons by virtue of their electronegativity. Therefore, in standard organic reactions, the majority of new bonds are formed between atoms of opposite polarity. This can be considered to be the "normal" mode of reactivity.
One consequence of this natural polarization of molecules is that 1,3- and 1,5- heteroatom substituted carbon skeletons are extremely easy to synthesize (Aldol reaction, Claisen condensation, Michael reaction, Claisen rearrangement, Diels-Alder reaction), whereas 1,2-, 1,4-, and 1,6- heteroatom substitution patterns are more difficult to access via "normal" reactivity. It is therefore important to understand and develop methods to induce umpolung in organic reactions.
Examples edit
The simplest method of obtaining 1,2-, 1,4-, and 1,6- heteroatom substitution patterns is to start with them. Biochemical and industrial processes can provide inexpensive sources of chemicals that have normally inaccessible substitution patterns. For example, amino acids, oxalic acid, succinic acid, adipic acid, tartaric acid, and glucose are abundant and provide nonroutine substitution patterns.
Cyanide-type umpolung edit
The canonical umpolung reagent is the cyanide ion. The cyanide ion is unusual in that a carbon triply bonded to a nitrogen would be expected to have a (+) polarity due to the higher electronegativity of the nitrogen atom. Yet, the negative charge of the cyanide ion is localized on the carbon, giving it a (-) formal charge. This chemical ambivalence results in umpolung in many reactions where cyanide is involved.
For example, cyanide is a key catalyst in the benzoin condensation, a classical example of polarity inversion.

The net result of the benzoin reaction is that a bond has been formed between two carbons that are normally electrophiles.
N-heterocyclic carbenes edit

N-heterocyclic carbenes are similar to cyanide in reactivity. Like cyanide, they have an unusual chemical ambivalence, which allows them to trigger umpolung in reactions where they are involved. The carbene has six electrons - two each in the carbon-nitrogen single bonds, two in its sp2-hybridized orbital, and an empty p-orbital. The sp2 lone pair acts as an electron donor, whereas the empty p-orbital is capable as acting as an electron acceptor.
In this example, the β-carbon of the α,β-unsaturated ester 1 formally acts as a nucleophile,[4] whereas normally it would be expected to be a Michael acceptor.

This carbene reacts with the α,β-unsaturated ester 1 at the β-position forming the intermediate enolate 2. Through tautomerization 2b can displace the terminal bromine atom to 3. An elimination reaction regenerates the carbene and releases the product 4.
For comparison: in the Baylis-Hillman reaction the same electrophilic β-carbon atom is attacked by a reagent but resulting in the activation of the α-position of the enone as the nucleophile.
Thiamine pyrophosphate edit
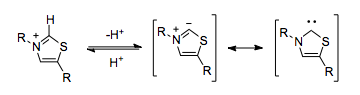
Biological processes can employ cyanide-like umpolung reactivity without having to rely on the toxic cyanide ion. Thiamine (which itself is an N-heterocyclic carbene) pyrophosphate (TPP) serves a functionally identical role. The thiazolium ring in TPP is deprotonated within the hydrophobic core of the enzyme,[5] resulting in a carbene which is capable of umpolung.
Enzymes which use TPP as a cofactor can catalyze umpolung reactivity, such as the decarboxylation of pyruvate.
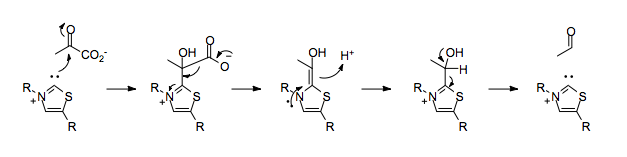
In the absence of TPP, the decarboxylation of pyruvate would result in the placement of a negative charge on the carbonyl carbon, which would run counter to the normal polarization of the carbon-oxygen double bond.
3-membered rings edit
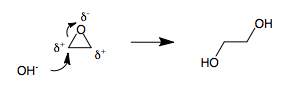
3-membered rings are strained moieties in organic chemistry. When a 3-membered ring contains a heteroatom, such as in an epoxide or in a bromonium intermediate, the three atoms in the ring become polarized. It is impossible to assign (+) and (-) polarities to a 3-membered ring without having two adjacent atoms with the same polarity. Therefore, whenever a polarized 3-membered ring is opened by a nucleophile, umpolung inevitably results .[citation needed] For example, the opening of ethylene oxide with hydroxide leads to ethylene glycol.
Carbonyl umpolung / anion relay chemistry edit
Dithiane chemistry is a classic example of polarity inversion. This can be observed in the Corey-Seebach reaction.
Ordinarily the oxygen atom in the carbonyl group is more electronegative than the carbon atom and therefore the carbonyl group reacts as an electrophile at carbon. This polarity can be reversed when the carbonyl group is converted into a dithiane or a thioacetal. In synthon terminology the ordinary carbonyl group is an acyl cation and the dithiane is a masked acyl anion.
When the dithiane is derived from an aldehyde such as acetaldehyde the acyl proton can be abstracted by n-butyllithium in THF at low temperatures. The thus generated 2-lithio-1,3-dithiane reacts as a nucleophile in nucleophilic displacement with alkyl halides such as benzyl bromide, with other carbonyl compounds such as cyclohexanone or oxiranes such as phenyl-epoxyethane, shown below. After hydrolysis of the dithiane group the final reaction products are α-alkyl-ketones or α-hydroxy-ketones. A common reagent for dithiane hydrolysis is (bis(trifluoroacetoxy)iodo)benzene.

Dithiane chemistry opens the way to many new chemical transformations. One example is found in so-called anion relay chemistry in which a negative charge of an anionic functional group resulting from one organic reaction is transferred to a different location within the same carbon framework and available for secondary reaction.[6] In this example of a multi-component reaction both formaldehyde (1) and isopropylaldehyde (8) are converted into dithianes 3 and 9 with 1,3-propanedithiol. Sulfide 3 is first silylated by reaction with tert-butyllithium and then trimethylsilyl chloride 4 and then the second acyl proton is removed and reacted with optically active (−)-epichlorohydrin 6 replacing chlorine. This compound serves as the substrate for reaction with the other dithiane 9 to the oxirane ring opening product 10. Under influence of the polar base HMPA, 10 rearranges in a 1,4-Brook rearrangement to the silyl ether 11 reactivating the formaldehyde dithiane group as an anion (hence the anion relay concept). This dithiane group reacts with oxirane 12 to the alcohol 13 and in the final step the sulfide groups are removed with (bis(trifluoroacetoxy)iodo)benzene.
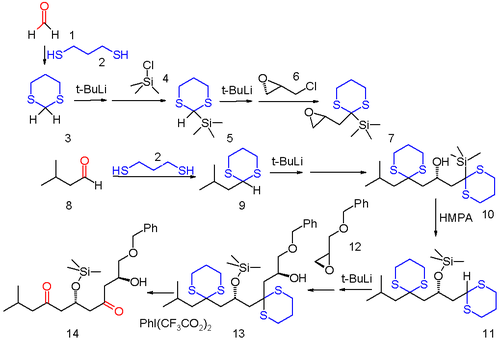
The anion relay chemistry tactic has been applied elegantly in the total synthesis of complex molecules of significant biological activity, such as spongistatin 2[7] and mandelalide A.[8][9]
Oxidative bond formation edit
It is possible to form a bond between two carbons of (-) polarity by using an oxidant such as iodine. In this total synthesis of enterolactone,[10] the 1,4- relationship of oxygen substituents is assembled by the oxidative homocoupling of a carboxylate enolate using iodine as the oxidant.

Amine umpolung edit
Ordinarily the nitrogen atom in the amine group is reacting as a nucleophile by way of its lone pair. This polarity can be reversed when a primary or secondary amine is substituted with a good leaving group (such as a halogen atom or an alkoxy group). The resulting N-substituted compound can behave as an electrophile at the nitrogen atom and react with a nucleophile as for example in the electrophilic amination of carbanions.[11]
Hydrazone umpolung edit
Recently, various carbonyls have been turned into organometallic reagent surrogates via hydrazone umpolung by C.-J. Li et al. In the presence of a catalyst, similar to organometallic reagents, hydrazones can undergo nucleophilic additions, conjugate additions, and transition-metal catalyzed cross-couplings with various electrophiles to form new C-C bonds.[12]
References edit
- ^ Seebach, D. (1979). "Methods of Reactivity Umpolung". Angewandte Chemie International Edition in English. 18 (4): 239–258. doi:10.1002/anie.197902393.
- ^ Gröbel, B. T.; Seebach, D. (1977). "Umpolung of the Reactivity of Carbonyl Compounds Through Sulfur-Containing Reagents". Synthesis. 1977 (6): 357. doi:10.1055/s-1977-24412. S2CID 95705677.
- ^ Seebach, D.; Corey, E. J. (1975). "Generation and synthetic applications of 2-lithio-1,3-dithianes". The Journal of Organic Chemistry. 40 (2): 231. doi:10.1021/jo00890a018.
- ^ Fischer, C.; Smith, S. W.; Powell, D. A.; Fu, G. C. (2006). "Umpolung of Michael Acceptors Catalyzed by N-Heterocyclic Carbenes". Journal of the American Chemical Society. 128 (5): 1472–1473. doi:10.1021/ja058222q. PMC 2553003. PMID 16448117.
- ^ Washabaugh, M. W.; Jencks, W. P. (1988). "Thiazolium C(2)-proton exchange: Structure-reactivity correlations and the pKa of thiamin C(2)-H revisited". Biochemistry. 27 (14): 5044–5053. doi:10.1021/bi00414a015. PMID 2844248.
- ^ Smith A. B. , III, Xian M. (2006). "Anion Relay Chemistry: An Effective Tactic for Diversity Oriented Synthesis". Journal of the American Chemical Society. 128 (1): 66–67. doi:10.1021/ja057059w. PMID 16390124.
{{cite journal}}: CS1 maint: multiple names: authors list (link) - ^ Smith A. B. , III., Lin Q., Doughty V. A., Zhuang L., McBriar M. D., Kerns J. K., Brook C. S., Murase N.,Nakayama K. (2001). "The Spongistatins: Architecturally Complex Natural Products—Part Two: Synthesis of the C(29–51) Subunit, Fragment Assembly, and Final Elaboration to (+)-Spongistatin 2". Angewandte Chemie International Edition. 40 (1): 196–199. doi:10.1002/1521-3773(20010105)40:1<196::AID-ANIE196>3.0.CO;2-T. PMID 11169711.
{{cite journal}}: CS1 maint: multiple names: authors list (link) - ^ Nguyen M. H., Imanishi M., Kurogi T., Smith A. B. , III. (2016). "Total Synthesis of (−)-Mandelalide A Exploiting Anion Relay Chemistry (ARC): Identification of a Type II ARC/CuCN Cross-Coupling Protocol". Journal of the American Chemical Society. 138 (11): 3675–3678. doi:10.1021/jacs.6b01731. PMC 4819492. PMID 26954306.
{{cite journal}}: CS1 maint: multiple names: authors list (link) - ^ Nguyen M. H., Imanishi M., Kurogi T., Wan, X., Ishmael, J., McPhail, K., Smith A. B. , III. (2018). "Synthetic Access to the Mandelalide Family of Macrolides: Development of an Anion Relay Chemistry Strategy". The Journal of Organic Chemistry. 83 (8): 4287–4306. doi:10.1021/acs.joc.8b00268. PMC 5910188. PMID 29480727.
{{cite journal}}: CS1 maint: multiple names: authors list (link) - ^ Belletire, J.L.; Fremont, S.L. (1986). "Oxidative coupling". Tetrahedron Letters. 27 (2): 127. doi:10.1016/S0040-4039(00)83958-1.
- ^ Erdik, E.; Ay, M. (1989). "Electrophilic amination of carbanions". Chemical Reviews. 89 (8): 1947–1980. doi:10.1021/cr00098a014.
- ^ Dai, X.J.; Li, C.C.; Li, C.-J. (2021). "Carbonyl umpolung as an organometallic reagent surrogate". Chemical Society Reviews. 50 (19): 10733–10742. doi:10.1039/D1CS00418B. PMID 34382626. S2CID 236989985.
External links edit
- IUPAC, Compendium of Chemical Terminology, 2nd ed. (the "Gold Book") (1997). Online corrected version: (2006–) "umpolung". doi:10.1351/goldbook.U06551