


Summary
Underrepresented populations, especially black and hispanic populations with cystic fibrosis are often not successfully diagnosed.[1] This is in part due to the minimal dissemination of existing data on patients from these underrepresented groups. While white populations do appear to experience a higher frequency of cystic fibrosis, other ethnicities are also affected and not always by the same biological mechanisms.[2] Thus, many healthcare and treatment options are less reliable or unavailable to underrepresented populations. This issue affects the level at which public health needs are being met across the world.

Definition edit
Cystic fibrosis (CF) is an autosomal recessive and monogenetic disorder. It is caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene.[3] The CFTR protein (Figure 1) serves to move chloride ions to the surface of cells to ensure proper hydration. When this protein becomes dysfunctional, the chloride ions are not present to thin out the viscous mucus that forms. This thickened mucus causes many problems in various organs of the body.[4]
Symptoms edit

People with cystic fibrosis may experience salty skin, persistent coughing, lung infections such as pneumonia and bronchitis, and wheezing and shortness of breath. Cystic fibrosis can also cause poor weight gain and growth, nasal polyps, chronic sinus infections, clubbing or enlargement of fingers and toes, infertility in males, and rectal prolapse.[4] Cystic fibrosis may also lead to arthropathies.[5] These symptoms and co-morbidities can ultimately lead to nutritional deficits and highly decreased quality of life.[6]
Compared to white patients, black patients have more severe pulmonary imaging findings and display more respiratory symptoms at diagnosis. Similarly, black and hispanic patients have overall worse pulmonary function than white patients that is often present earlier in life.[7] Though this may be the case, these symptoms can be misdiagnosed as other health issues and treated as such.[8]
Diagnosis edit
Through symptoms edit
To be diagnosed with CF, patients must meet certain criteria. These criteria may include showing a combination of the signs and symptoms above and/or have a family member with CF and/or test positive for the mutation during genetic screening.
Conventional diagnostic criteria edit
Individuals must have at least one clinical feature:[3]
- meconium ileus (bowel obstruction when meconium in intestine is too thick)
- diarrhea and failure to thrive
- recurrent respiratory infections
- nasal polyps
- rectal prolapse
- male infertility
- electrolyte depletion
Or CF diagnosis in family member, or positive newborn screening test, plus one or both of the following:
- chloride channel dysfunction (positive sweat test or abnormal transepithelial potential difference)
- known disease-causing mutations on chromosome 7 in trans
Though these symptoms may present themselves in underrepresented minorities, there remains the misconception among doctors that underrepresented minorities rarely have the disease.[9] This results in under-diagnosis in these populations. Also, in developing countries, such as in Africa, where HIV, protein energy malnutrition,[10] and chronic pulmonary infections are prominent, it can be difficult for clinicians to rightly diagnose patients with CF.[1][8] Proper diagnosis can also be affected when patients show a positive test but do not display symptoms or display symptoms but do not show positive diagnostic tests. For this reason, it is very important to continue to monitor patients in both categories, especially those of the latter while treating the symptoms that may be disguised as another disease.[3]
Through screening edit
In many western countries, prenatal and newborn genetic screening is available. This has been correlated with decreased incidence as patients and their families are able to seek treatment early on.[3][11] These screening methods may include measuring serum immunoreactive trypsin (iRT) and CFTR mutation analysis.[12][13]
CFTR mutation classification systems typically include six classes[14] where classes I-III tend to have more severe symptoms than classes IV-VI.[1]
CFTR mutation classification edit
- Protein production mutations (Class I)
- Protein processing mutations (Class II)
- Gating mutations (Class III)
- Conduction mutations (Class IV)
- Insufficient CFTR transcript mutations (Class V)
- Reduced protein stability and increased turnover of CFTR at the cell surface (Class VI)
IRT and CFTR mutation analyses are always confirmed with what is known as a sweat test which will show elevated levels of chloride in sweat of CF patients.[3][15] However, this is not a perfect system and many children end up with an inconclusive diagnosis and need to be monitored for symptoms that may not show up until much later in life.[16] Also, in some states such as New Jersey, there is only one mutation on the CFTR screening panel (F508del). Because 12.9% of CF patients in the United States do not have this specific mutation, they cannot be diagnosed using this particular screening method. This value can be even larger when narrowing in on minority populations (see race and ethnicity).[8][17] Upon analysis of the 2020 Cystic Fibrosis Foundation Patient Registry, the detection rate of CFTR mutation variants in known CF patients was highest in white patients at a false negative rate of only 3–5%.[11]
| Race / ethnicity | Detection rate (%) | Detection failure rate (%) |
|---|---|---|
| Asian | 56-77 | 23-44 |
| Black | 73-86 | 14-27 |
| American Indian and Alaskan Natives | 84-91 | 9-16 |
| Hispanic | 81-94 | 6-19 |
| White | 95-97 | 3-5 |
Risk factors, incidence, and prevalence edit
Genetics, sex, and age edit
Since the CFTR mutation is autosomal recessive, the most significant risk factor is family history. The disease only manifests itself when a child inherits CFTR mutations from both parents. Children who inherit a mutation from one parent are considered carriers and can pass it on to their children.[4] With this being the case, mutations may be family and region specific (See Race, ethnicity, and geographic location).[17] In regard to sex, CF occurs in occurs in males and females; however, women are more likely to display worse symptoms and outcomes.[18] This is particularly relevant in regard to Peudomonas aeruginosa[19] respiratory infections.[20] Age-wise, most people are diagnosed by the age of 2; however, the Cystic Fibrosis Foundation Patient Registry shows that more than half of the CF population is age 18 or older in the United States.[4]
Race, ethnicity, and geographic location edit
Cystic fibrosis occurs in all races, but may be most prevalent in white people of Northern European ancestry.[21] CF incidence in other populations may be underreported as there are hundreds of CFTR mutations that can manifest the disease and not all have been identified. Though there has been a decrease in incidence in more developed countries due to prenatal genetic screening,[22][23] the prevalence is expected to increase as people are able to live longer with the disease.[3]
F508del is the most common mutation across all cystic fibrosis patients.[24] However, it is not the most common mutation found in specific geographical locations.
| Frequency (%) | Location |
|---|---|
| 70 | central, northern, western, and northeastern Europe |
| 100 | Faroe Islands of Denmark |
| 20 | Turkey |
| 60 | Argentina and Uruguay |
| 40 | Brazil, Chile, Colombia, Mexico, and Venezuela |
| 20-30 | Puerto Rico, Cuba, Ecuador, and Costa Rica |
| 10 | Dominican Republic |
| 60 | Pakistan |
| 20 | India |
| 10 | Japan |
Since a popular method of identification is genetic testing for this particular mutation, the identification frequency is lower in underrepresented population.
| Identification category | Frequency (%) | Race |
|---|---|---|
| Unclassified | 25 | Black, Hispanic, and other races |
| 11 | non-Hispanic White | |
| One unidentified | 8-10 | Black, Hispanic, and other races |
| 3 | non-Hispanic White people |
African descendants display the most genetic diversity across the human population. For CF patients of African descent, this means that they are more likely to harbor less common CFTR mutations.[1] Some mutations that are unique to Africa are 2766del8, 1670delC, Y1109x, and A204T.

| Country | Mutations |
|---|---|
| Morocco | ΔF508, 5T, 12TG, 11TG, 711+1G>T, R74W, R1070W, D1270N, 3849+10kbC>T, S549R, G1244E, U |
| Algeria | ΔF508, 1609delCA, N1303K, 711+1G>T, 1812-1G>A, 5T, E1104X, U |
| Tunisia | ΔF508, W1282X, 711+1G>T, E1104X, R74W, Y122X, V201M, R1158X, 4016insT, U, G542X, N1303K, 405+1G>A, G85E, D1270N, R1066C, I1203V, R785X, 5T, 2766del8, F1166C, 3729delAinsTCT, 1811+5A>G, T665S, L1043R, 4268+2T>G |
| Libya | 1670delC, ΔF508, E1104X, N1303K, U |
| Egypt | ΔF508, N1303K, U, 1838+3A>C, T665S, 5T, 7T, 9T |
| Sudan | D579G, R1102K |
| Senegal | U, 4136+1G>A, EX17a-EX18del |
| Cameroon | Y1109x, 405+4A>G |
| Rwanda | F693L, T854T, M470V, P1290P, E527E, U, 3120+1G>A, Q1463Q, 1898+152T>A, 1001+11C>T, 2752-15C>G, A204T, 3041-71A>G, 4575+2G>A, 3272-32T>C |
| Namibia | ΔF508 |
| Zimbabwe | 3120+1G>A, c.54-1161_c.164+1603del2875 |
| South Africa | ΔF508, G1249E, D1270N, 394deITT, R553X, N1303K, R117H, S549N, 1717-1G>A, 3659deIC, 2183delAA, 3120+1G>A, 3196de154, 3272-26A>G, G542X, W1282X, G5510, 0493X, 621+1G>T, 278945G>A, R1162X, U, -94G>T, c.54-1161_c.164+1603del2875 |
Treatment edit
Treatment is most effective with early diagnosis. As CF is a complicated ailment, patients must often use a combination of therapies.[4]
Airway clearance edit
Clearing the airways of mucus can help decrease the incidence of infection in the lung and improve lung function. The most common airway clearance techniques include deep coughing, active breathing therapy, autogenic drainage, and positive expiratory pressure.[citation needed]
Inhaled medications edit
There are several available inhaled medications such as bronchodilators and mucus thinners. These are medications that are delivered as a mist or aerosol and inhaled through a nebulizer. They may also include antibiotics.[citation needed]

Antibiotics edit
Antibiotics are used to fight bacterial infections which CF patients are very likely to develop due to thickened mucus. They are often taken daily by CF patients. It is recommended that inhaled antibiotics be taken only when the airway has been clear so that the drug can reach the affected area easily.[citation needed]
Pancreatic enzyme supplements edit
Pancreatic enzyme supplements serve to improve pancreatic function by increasing the absorption of essential nutrients. They are taken with meals. Multivitamins as supplements are also recommended for CF patients.[25]
Fitness plans edit
A personalized fitness plan can aid in airway clearance, improve energy, increase lung function, and help the overall health of the patient. [26]
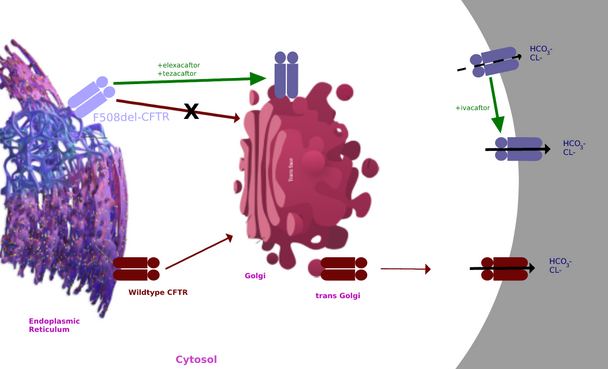
CFTR modulators edit
CFTR modulators target the mutated CFTR protein.[27][28] Thus far, there are some approved CFTR modulators for specific mutations. One such drug is elexacaftor/tezacaftor/ivacaftor. However, the cost of CFTR modulators is high.[29] In addition to the high cost, the available CFTR modulators may not be as effective in minority populations.[30] In fact, while 92.4% of white patients are eligible for treatment with CFTR modulators, only 69.7% and 75.6% of Hispanic patients are eligible.[30]
Global impact edit
Healthcare access edit
Communities with large black and hispanic populations are also most likely to have lower quality/less available healthcare.[31] This means that the risk of mistakes being made in screening and diagnosis are more likely to happen in these areas. The clinics and hospitals are often understaffed and rushing through samples can cause inaccurate screening results.[8] Furthermore, the cystic fibrosis screening tests for CFTR present false negative results most often in black and hispanic babies.[11][32] This happens because the CFTR variants most tested for are those that are largely found in white populations.[11] Also, as previously mentioned, one of the most common methods of diagnosis is using the sweat test. However, this test has a disproportionate rate of false negatives in areas with less healthcare access.[11][30] The presence of implicit biases in healthcare also contribute to this.[7][33] Additionally, availability of genetic screening options in developing countries and areas of lower socioeconomic status has proven to be difficult both because of financial and resource burden.[34] Altogether, this means that black and hispanic families are more likely to be diagnosed later on and therefore do not receive timely and as effective treatment.[citation needed]
Prognosis and quality of life in underrepresented populations edit
It is still up for debate on whether the overall prognosis of CF is improving. In areas where genetic screening is available, clinicians are able to identify and provide treatment for CF early on. In areas where this is not available, many patients are not diagnosed until later in life and display worse symptoms and poorer management of symptoms.[35] This is often in areas where there is a high density of underrepresented groups.[36] With the reduced access to healthcare, minority populations experience worse outcomes, even after taking low socioeconomic status into account.[7] Although hispanic patients are more likely than white patients to have milder CFTR mutations (those from Classes IV-VI), they suffer worse outcomes.
| Frequency (%) | Race |
|---|---|
| 75 | non-Hispanic White |
| 50 | Black, Hispanic, and other races |
Current programs and research edit
In addition to focusing on improving detection and diagnosis, understanding CF microorganisms, and developing new treatments, optimizing current treatments, and evaluating long-term antimicrobial use, the Cystic Fibrosis Foundation is currently[when?] striving for more equity and timeliness in cystic fibrosis newborn screening.[37] Cystic Fibrosis Research Institute has implemented strategies to increase awareness in underrepresented populations.[38] Though there is ongoing research about cystic fibrosis in underrepresented populations, many of the studies leave much to be desired and are not performed to the standards of studies conducted in white patients.[citation needed][editorializing]
References edit
- ^ a b c d e Stewart, Cheryl; Pepper, Michael S. (2016). "Cystic fibrosis on the African continent". Genetics in Medicine. 18 (7): 653–662. doi:10.1038/gim.2015.157. hdl:2263/56176. ISSN 1530-0366. PMID 26656651. S2CID 3675593.
- ^ Tishkoff, Sarah A.; Kidd, Kenneth K. (November 2004). "Implications of biogeography of human populations for 'race' and medicine". Nature Genetics. 36 (11): S21–S27. doi:10.1038/ng1438. ISSN 1546-1718. PMID 15507999. S2CID 1500915.
- ^ a b c d e f Ratjen, Felix; Bell, Scott C.; Rowe, Steven M.; Goss, Christopher H.; Quittner, Alexandra L.; Bush, Andrew (2015-05-14). "Cystic fibrosis". Nature Reviews. Disease Primers. 1: 15010. doi:10.1038/nrdp.2015.10. ISSN 2056-676X. PMC 7041544. PMID 27189798.
- ^ a b c d e "About Cystic Fibrosis". Cystic Fibrosis Foundation. Retrieved 2022-11-18.
- ^ Roehmel, Jobst F.; Kallinich, Tilmann; Staab, Doris; Schwarz, Carsten (2019-02-01). "Clinical manifestations and risk factors of arthropathy in cystic fibrosis". Respiratory Medicine. 147: 66–71. doi:10.1016/j.rmed.2019.01.003. ISSN 0954-6111. PMID 30704701. S2CID 73437549.
- ^ Patel, Dhiren; Shan, Albert; Mathews, Stacy; Sathe, Meghana (2022-02-28). "Understanding Cystic Fibrosis Comorbidities and Their Impact on Nutritional Management". Nutrients. 14 (5): 1028. doi:10.3390/nu14051028. ISSN 2072-6643. PMC 8912424. PMID 35268004.
- ^ a b c McGarry, Meghan E.; Williams, Wadsworth A.; McColley, Susanna A. (November 2019). "The Demographics of Adverse Outcomes in Cystic Fibrosis". Pediatric Pulmonology. 54 (Suppl 3): S74–S83. doi:10.1002/ppul.24434. ISSN 8755-6863. PMC 6857719. PMID 31715087.
- ^ a b c d Rock, Michael J.; Levy, Hara; Zaleski, Christina; Farrell, Philip M. (December 2011). "Factors Accounting for a Missed Diagnosis of Cystic Fibrosis After Newborn Screening". Pediatric Pulmonology. 46 (12): 1166–1174. doi:10.1002/ppul.21509. ISSN 8755-6863. PMC 4469987. PMID 22081556.
- ^ Mutesa, L.; Bours, V. (2009-07-22). "Diagnostic Challenges of Cystic Fibrosis in Patients of African Origin". Journal of Tropical Pediatrics. 55 (5): 281–286. doi:10.1093/tropej/fmp064. ISSN 0142-6338. PMID 19625487.
- ^ Schönfeldt, Hettie Carina; Gibson Hall, Nicolette (2012-02-22). "Dietary protein quality and malnutrition in Africa". British Journal of Nutrition. 108 (S2): S69–S76. doi:10.1017/S0007114512002553. hdl:2263/20602. ISSN 0007-1145. PMID 23107550.
- ^ a b c d e f Murez, Cara (October 31, 2022). "Cystic Fibrosis Screening Often Misses Black, Hispanic Babies". U.S. News HealthDay. University of California, San Francisco. Retrieved November 7, 2022.
- ^ Rock, Michael J.; Farrell, Philip M. (2006-01-01), Chernick, Victor; Boat, Thomas F.; Wilmott, Robert W.; Bush, Andrew (eds.), "Chapter 59 - Neonatal Screening for Cystic Fibrosis", Kendig's Disorders of the Respiratory Tract in Children (Seventh Edition), Philadelphia: W.B. Saunders, pp. 861–865, doi:10.1016/b978-0-7216-3695-5.50063-8, ISBN 978-0-7216-3695-5, retrieved 2022-12-09
- ^ Pletcher, Beth A. (2009-01-01), Turcios, Nelson L.; Fink, Robert J. (eds.), "Chapter 14 - Pulmonary Manifestations of Genetic Diseases", Pulmonary Manifestations of Pediatric Diseases, Philadelphia: W.B. Saunders, pp. 295–338, doi:10.1016/b978-1-4160-3031-7.00014-0, ISBN 978-1-4160-3031-7, retrieved 2022-12-09
- ^ Ikpa, Pauline T.; Bijvelds, Marcel J. C.; de Jonge, Hugo R. (2014-07-01). "Cystic fibrosis: Toward personalized therapies". The International Journal of Biochemistry & Cell Biology. Cystic Fibrosis: From o-mics to cell biology, physiology, and therapeutic advances. 52: 192–200. doi:10.1016/j.biocel.2014.02.008. ISSN 1357-2725. PMID 24561283.
- ^ Gokdemir, Yasemin; Karadag, Bulent Taner (2021). "Sweat Testing and Recent Advances". Frontiers in Pediatrics. 9: 649904. doi:10.3389/fped.2021.649904. ISSN 2296-2360. PMC 8129525. PMID 34017807.
- ^ Munck, Anne (2020-03-12). "Inconclusive Diagnosis after Newborn Screening for Cystic Fibrosis". International Journal of Neonatal Screening. 6 (1): 19. doi:10.3390/ijns6010019. ISSN 2409-515X. PMC 7422971. PMID 33073016.
- ^ a b c d e McGarry, Meghan E.; Gibb, Elizabeth R.; Oates, Gabriela R.; Schechter, Michael S. (2022-06-01). "Left behind: The potential impact of CFTR modulators on racial and ethnic disparities in cystic fibrosis". Paediatric Respiratory Reviews. 42: 35–42. doi:10.1016/j.prrv.2021.12.001. ISSN 1526-0542. PMC 9356388. PMID 35277357.
- ^ Healey, Natalie (2020-07-29). "The gender gap in cystic fibrosis". Nature. 583 (7818): S10–S11. Bibcode:2020Natur.583S..10H. doi:10.1038/d41586-020-02110-0. S2CID 220843285.
- ^ D'Agata, Erika (2015-01-01), Bennett, John E.; Dolin, Raphael; Blaser, Martin J. (eds.), "221 - Pseudomonas aeruginosa and Other Pseudomonas Species", Mandell, Douglas, and Bennett's Principles and Practice of Infectious Diseases (Eighth Edition), Philadelphia: W.B. Saunders, pp. 2518–2531.e3, ISBN 978-1-4557-4801-3, retrieved 2022-12-09
- ^ Harness-Brumley, Cayce L.; Elliott, Alan C.; Rosenbluth, Daniel B.; Raghavan, Deepa; Jain, Raksha (2014-12-01). "Gender Differences in Outcomes of Patients with Cystic Fibrosis". Journal of Women's Health. 23 (12): 1012–1020. doi:10.1089/jwh.2014.4985. ISSN 1540-9996. PMC 4442553. PMID 25495366.
- ^ "Cystic fibrosis - Symptoms and causes". Mayo Clinic. Retrieved 2022-11-18.
- ^ "CF Foundation Estimates Increase in CF Population". Cystic Fibrosis Foundation. 28 July 2022. Retrieved 2022-11-18.
- ^ Scotet, Virginie; L'Hostis, Carine; Férec, Claude (2020-05-26). "The Changing Epidemiology of Cystic Fibrosis: Incidence, Survival and Impact of the CFTR Gene Discovery". Genes. 11 (6): 589. doi:10.3390/genes11060589. ISSN 2073-4425. PMC 7348877. PMID 32466381.
- ^ "Featured Review: Drugs for correcting the basic defect in the most common cystic fibrosis-causing gene variant". Cochrane. Retrieved 2022-11-18.
- ^ Borowitz, Drucy; Wegman, Theresa; Harris, Margaret (December 1994). "Preventive Care for Patients With Chronic Illness: Multivitamin Use in Patients With Cystic Fibrosis". Clinical Pediatrics. 33 (12): 720–725. doi:10.1177/000992289403301203. ISSN 0009-9228. PMID 7874824. S2CID 43047336. Retrieved 10 December 2022.
- ^ Ward, Nathan; Morrow, Scott; Stiller, Kathy; Holland, Anne E. (1 August 2021). "Exercise as a substitute for traditional airway clearance in cystic fibrosis: a systematic review". Thorax. 76 (8): 763–771. doi:10.1136/thoraxjnl-2020-215836. ISSN 0040-6376. PMID 33443204. S2CID 229357310. Retrieved 10 December 2022.
- ^ Habib, Al-Rahim R.; Kajbafzadeh, Majid; Desai, Sameer; Yang, Connie L.; Skolnik, Kate; Quon, Bradley S. (2019-05-10). "A Systematic Review of the Clinical Efficacy and Safety of CFTR Modulators in Cystic Fibrosis". Scientific Reports. 9 (1): 7234. Bibcode:2019NatSR...9.7234H. doi:10.1038/s41598-019-43652-2. ISSN 2045-2322. PMC 6510767. PMID 31076617.
- ^ Lopes-Pacheco, Miquéias (2020). "CFTR Modulators: The Changing Face of Cystic Fibrosis in the Era of Precision Medicine". Frontiers in Pharmacology. 10: 1662. doi:10.3389/fphar.2019.01662. ISSN 1663-9812. PMC 7046560. PMID 32153386.
- ^ Goetz, Danielle M.; Savant, Adrienne P. (December 2021). "Review of CFTR modulators 2020". Pediatric Pulmonology. 56 (12): 3595–3606. doi:10.1002/ppul.25627. ISSN 8755-6863. PMID 34407318. S2CID 237216168.
- ^ a b c McGarry, Meghan E.; McColley, Susanna A. (June 2021). "Cystic Fibrosis Patients of Minority Race and Ethnicity Less Likely Eligible for CFTR Modulators Based on CFTR Genotype". Pediatric Pulmonology. 56 (6): 1496–1503. doi:10.1002/ppul.25285. ISSN 8755-6863. PMC 8137541. PMID 33470563.
- ^ "Racism, Inequality, and Health Care for African Americans". The Century Foundation. 2019-12-19. Retrieved 2022-12-09.
- ^ Rho, Jason; Ahn, Chul; Gao, Ang; Sawicki, Gregory S.; Keller, Ashley; Jain, Raksha (2018-10-15). "Disparities in Mortality of Hispanic Patients with Cystic Fibrosis in the United States. A National and Regional Cohort Study". American Journal of Respiratory and Critical Care Medicine. 198 (8): 1055–1063. doi:10.1164/rccm.201711-2357OC. ISSN 1073-449X. PMC 6221571. PMID 29742360.
- ^ "Implicit Bias and Racial Disparities in Health Care". www.americanbar.org. Retrieved 2022-11-18.
- ^ da Silva Filho, Luiz Vicente Ribeiro Ferreira; Zampoli, Marco; Cohen-Cymberknoh, Malena; Kabra, Sushil K. (2021-06-01). "Cystic fibrosis in low and middle-income countries (LMIC): A view from four different regions of the world". Paediatric Respiratory Reviews. 38: 37–44. doi:10.1016/j.prrv.2020.07.004. ISSN 1526-0542. PMID 32826173. S2CID 221240158.
- ^ DiMango, Emily; Simpson, Kaitlyn; Menten, Elizabeth; Keating, Claire; Fan, Weijia; Leu, Cheng-Shiun (2021-07-31). "Health Disparities among adults cared for at an urban cystic fibrosis program". Orphanet Journal of Rare Diseases. 16 (1): 332. doi:10.1186/s13023-021-01965-4. ISSN 1750-1172. PMC 8325847. PMID 34332588.
- ^ McBennett, Kimberly A.; Davis, Pamela B.; Konstan, Michael W. (February 2022). "Increasing life expectancy in cystic fibrosis: Advances and challenges". Pediatric Pulmonology. 57 (Suppl 1): S5–S12. doi:10.1002/ppul.25733. ISSN 8755-6863. PMC 9004282. PMID 34672432.
- ^ "CF Foundation Continues Working Toward Equity and Timeliness in Cystic Fibrosis Newborn Screening". Cystic Fibrosis Foundation. 29 September 2022. Retrieved 2022-11-18.
- ^ "Diversity and Inclusion". Cystic Fibrosis Research Institute. 17 December 2021. Retrieved 2022-11-18.