


Summary
MT-ATP8 (or ATP8) is a mitochondrial gene with the full name 'mitochondrially encoded ATP synthase membrane subunit 8' that encodes a subunit of mitochondrial ATP synthase, ATP synthase Fo subunit 8 (or subunit A6L). This subunit belongs to the Fo complex of the large, transmembrane F-type ATP synthase.[3] This enzyme, which is also known as complex V, is responsible for the final step of oxidative phosphorylation in the electron transport chain. Specifically, one segment of ATP synthase allows positively charged ions, called protons, to flow across a specialized membrane inside mitochondria. Another segment of the enzyme uses the energy created by this proton flow to convert a molecule called adenosine diphosphate (ADP) to ATP.[4] Subunit 8 differs in sequence between Metazoa, plants and Fungi.
| ATP8 | |||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Identifiers | |||||||||||||||||||||||||||||||||||||||||||||||||||
| Aliases | ATP8, ATPase8, MTMT-ATP synthase F0 subunit 8 | ||||||||||||||||||||||||||||||||||||||||||||||||||
| External IDs | OMIM: 516070 HomoloGene: 124425 GeneCards: ATP8 | ||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| Wikidata | |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||


| ATP synthase protein 8 (metazoa) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Identifiers | |||||||||
| Symbol | ATP-synt_8 | ||||||||
| Pfam | PF00895 | ||||||||
| Pfam clan | CL0255 | ||||||||
| InterPro | IPR001421 | ||||||||
| |||||||||
| Plant ATP synthase F0 subunit 8 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Identifiers | |||||||||
| Symbol | YMF19 | ||||||||
| Pfam | PF02326 | ||||||||
| Pfam clan | CL0255 | ||||||||
| InterPro | IPR003319 | ||||||||
| |||||||||
| Fungal ATP synthase protein 8 (A6L) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Identifiers | |||||||||
| Symbol | Fun_ATP-synt_8 | ||||||||
| Pfam | PF05933 | ||||||||
| Pfam clan | CL0255 | ||||||||
| InterPro | IPR009230 | ||||||||
| |||||||||
Structure edit
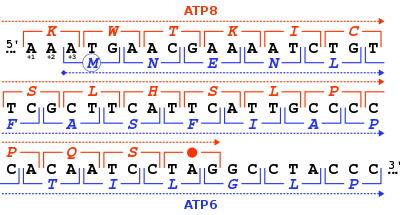
The ATP synthase protein 8 of human and other mammals is encoded in the mitochondrial genome by the MT-ATP8 gene. When the complete human mitochondrial genome was first published, the MT-ATP8 gene was described as the unidentified reading frame URF A6L.[3] An unusual feature of the MT-ATP8 gene is its 46-nucleotide overlap with the MT-ATP6 gene. With respect to the reading frame (+1) of MT-ATP8, the MT-ATP6 gene starts on the +3 reading frame.
The MT-ATP8 protein weighs 8 kDa and is composed of 68 amino acids.[5][6] The protein is a subunit of the F1Fo ATPase, also known as Complex V, which consists of 14 nuclear- and 2 mitochondrial-encoded subunits. F-type ATPases consist of two structural domains, F1 containing the extramembraneous catalytic core and Fo containing the membrane proton channel, linked together by a central stalk and a peripheral stalk. As an A subunit, MT-ATP8 is contained within the non-catalytic, transmembrane Fo portion of the complex, comprising the proton channel. The catalytic portion of mitochondrial ATP synthase consists of 5 different subunits (alpha, beta, gamma, delta, and epsilon) assembled with a stoichiometry of 3 alpha, 3 beta, and a single representative of the other 3. The proton channel consists of three main subunits (a, b, c). This gene encodes the delta subunit of the catalytic core. Alternatively spliced transcript variants encoding the same isoform have been identified.[7][4]
Function edit
The MT-ATP8 gene encodes a subunit of mitochondrial ATP synthase, located within the thylakoid membrane and the inner mitochondrial membrane. Mitochondrial ATP synthase catalyzes ATP synthesis, utilizing an electrochemical gradient of protons across the inner membrane during oxidative phosphorylation.[7] The Fo region causes rotation of F1, which has a water-soluble component that hydrolyzes ATP and together, the F1Fo creates a pathway for movement of protons across the membrane.[8]
This protein subunit appears to be an integral component of the stator stalk in yeast mitochondrial F-ATPases.[9] The stator stalk is anchored in the membrane, and acts to prevent futile rotation of the ATPase subunits relative to the rotor during coupled ATP synthesis/hydrolysis. This subunit may have an analogous function in Metazoa.
Nomenclature edit
The nomenclature of the enzyme has a long history. The F1 fraction derives its name from the term "Fraction 1" and Fo (written as a subscript letter "o", not "zero") derives its name from being the binding fraction for oligomycin, a type of naturally-derived antibiotic that is able to inhibit the Fo unit of ATP synthase.[10][11] The Fo region of ATP synthase is a proton pore that is embedded in the mitochondrial membrane. It consists of three main subunits A, B, and C, and (in humans) six additional subunits, d, e, f, g, MT-ATP6 (or F6), and MT-ATP8 (or A6L). 3D structure of E. coli homologue of this subunit was modeled based on electron microscopy data (chain M of PDB: 1c17). It forms a transmembrane 4-α-bundle.
Clinical Significance edit
Mutations to MT-ATP8 and other genes affecting oxidative phosphorylation in the mitochondria have been associated with a variety of neurodegenerative and cardiovascular disorders, including mitochondrial complex V deficiency, Leber's hereditary optic neuropathy (LHON), mitochondrial encephalomyopathy with stroke-like episodes (MELAS), Leigh syndrome, and NARP syndrome. Most of the body's cells contain thousands of mitochondria, each with one or more copies of mitochondrial DNA. The severity of some mitochondrial disorders is associated with the percentage of mitochondria in each cell that has a particular genetic change. People with Leigh syndrome due to a MT-ATP6 gene mutation tend to have a very high percentage of mitochondria with the mutation (from more than 90 percent to 95 percent). The less-severe features of NARP result from a lower percentage of mitochondria with the mutation, typically 70 percent to 90 percent. Because these two conditions result from the same genetic changes and can occur in different members of a single family, researchers believe that they may represent a spectrum of overlapping features instead of two distinct syndromes.[4]
Mitochondrial complex V deficiency presents with heterogeneous clinical manifestations including neuropathy, ataxia, hypertrophic cardiomyopathy. Hypertrophic cardiomyopathy can present with negligible to extreme hypertrophy, minimal to extensive fibrosis and myocyte disarray, absent to severe left ventricular outflow tract obstruction, and distinct septal contours/morphologies with extremely varying clinical course.[12][13]
Mitochondrial complex V deficiency is a shortage (deficiency) or loss of function in complex V of the electron transport chain that can cause a wide variety of signs and symptoms affecting many organs and systems of the body, particularly the nervous system and the heart. The disorder can be life-threatening in infancy or early childhood. Affected individuals may have feeding problems, slow growth, low muscle tone (hypotonia), extreme fatigue (lethargy), and developmental delay. They tend to develop elevated levels of lactic acid in the blood (lactic acidosis), which can cause nausea, vomiting, weakness, and rapid breathing. High levels of ammonia in the blood (hyperammonemia) can also occur in affected individuals, and in some cases result in abnormal brain function (encephalopathy) and damage to other organs.[14] Ataxia, microcephaly, developmental delay and intellectual disability have been observed in patients with a frameshift mutation in MT-ATP6. This causes a C insertion at position 8612 that results in a truncated protein only 36 amino acids long, and two T > C single-nucleotide polymorphisms at positions 8610 and 8614 that result in a homopolymeric cytosine stretch.[15]
Hypertrophic cardiomyopathy, a common feature of mitochondrial complex V deficiency, is characterized by thickening (hypertrophy) of the cardiac muscle that can lead to heart failure.[14] The m.8528T>C mutation occurs in the overlapping region of the MT-ATP6 and MT-ATP8 genes and has been described in multiple patients with infantile cardiomyopathy. This mutation changes the initiation codon in MT-ATP6 to threonine as well as a change from tryptophan to arginine at position 55 of MT-ATP8.[16][13] Individuals with mitochondrial complex V deficiency may also have a characteristic pattern of facial features, including a high forehead, curved eyebrows, outside corners of the eyes that point downward (downslanting palpebral fissures), a prominent bridge of the nose, low-set ears, thin lips, and a small chin (micrognathia).[14]
Infantile hypertrophic cardiomyopathy (CMHI) is also caused by mutations affecting distinct genetic loci, including MT-ATP6 and MT-ATP8. An infantile form of hypertrophic cardiomyopathy, a heart disorder characterized by ventricular hypertrophy, which is usually asymmetric and often involves the interventricular septum. The symptoms include dyspnea, syncope, collapse, palpitations, and chest pain. They can be readily provoked by exercise. The disorder has inter- and intrafamilial variability ranging from benign to malignant forms with high risk of cardiac failure and sudden cardiac death.[12][13]
References edit
- ^ a b c GRCh38: Ensembl release 89: ENSG00000228253 – Ensembl, May 2017
- ^ "Human PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- ^ a b Anderson S, Bankier AT, Barrell BG, de Bruijn MH, Coulson AR, Drouin J, Eperon IC, Nierlich DP, Roe BA, Sanger F, Schreier PH, Smith AJ, Staden R, Young IG (April 1981). "Sequence and organization of the human mitochondrial genome". Nature. 290 (5806): 457–65. Bibcode:1981Natur.290..457A. doi:10.1038/290457a0. PMID 7219534. S2CID 4355527.
- ^ a b c "MT-ATP8". Genetics Home Reference. NCBI.
- ^ Zong NC, Li H, Li H, Lam MP, Jimenez RC, Kim CS, Deng N, Kim AK, Choi JH, Zelaya I, Liem D, Meyer D, Odeberg J, Fang C, Lu HJ, Xu T, Weiss J, Duan H, Uhlen M, Yates JR, Apweiler R, Ge J, Hermjakob H, Ping P (Oct 2013). "Integration of cardiac proteome biology and medicine by a specialized knowledgebase". Circulation Research. 113 (9): 1043–53. doi:10.1161/CIRCRESAHA.113.301151. PMC 4076475. PMID 23965338.
- ^ "ATP synthase protein 8". Cardiac Organellar Protein Atlas Knowledgebase (COPaKB).[permanent dead link]
- ^ a b "MT-ATP8 mitochondrially encoded ATP synthase 8 [Homo sapiens (human)]". Gene. NCBI.
- ^ Velours J, Paumard P, Soubannier V, Spannagel C, Vaillier J, Arselin G, Graves PV (May 2000). "Organisation of the yeast ATP synthase F(0):a study based on cysteine mutants, thiol modification and cross-linking reagents". Biochimica et Biophysica Acta (BBA) - Bioenergetics. 1458 (2–3): 443–56. doi:10.1016/S0005-2728(00)00093-1. PMID 10838057.
- ^ Stephens AN, Khan MA, Roucou X, Nagley P, Devenish RJ (May 2003). "The molecular neighborhood of subunit 8 of yeast mitochondrial F1F0-ATP synthase probed by cysteine scanning mutagenesis and chemical modification". The Journal of Biological Chemistry. 278 (20): 17867–75. doi:10.1074/jbc.M300967200. PMID 12626501.
- ^ Kagawa Y, Racker E (May 1966). "Partial resolution of the enzymes catalyzing oxidative phosphorylation. 8. Properties of a factor conferring oligomycin sensitivity on mitochondrial adenosine triphosphatase". The Journal of Biological Chemistry. 241 (10): 2461–6. doi:10.1016/S0021-9258(18)96640-8. PMID 4223640.
- ^ Mccarty RE (November 1992). "A PLANT BIOCHEMIST'S VIEW OF H+-ATPases AND ATP SYNTHASES". The Journal of Experimental Biology. 172 (Pt 1): 431–441. doi:10.1242/jeb.172.1.431. PMID 9874753.
- ^ a b "MT-ATP8 - ATP synthase protein 8 - Homo sapiens (Human)". www.uniprot.org. UniProt. Retrieved 3 August 2018.
 This article incorporates text available under the CC BY 4.0 license.
This article incorporates text available under the CC BY 4.0 license.
- ^ a b c Ware SM, El-Hassan N, Kahler SG, Zhang Q, Ma YW, Miller E, Wong B, Spicer RL, Craigen WJ, Kozel BA, Grange DK, Wong LJ (May 2009). "Infantile cardiomyopathy caused by a mutation in the overlapping region of mitochondrial ATPase 6 and 8 genes". Journal of Medical Genetics. 46 (5): 308–14. doi:10.1136/jmg.2008.063149. PMID 19188198. S2CID 25354118.
- ^ a b c "Mitochondrial complex V deficiency". Genetics Home Reference. NCBI. Retrieved 3 August 2018.
 This article incorporates text from this source, which is in the public domain.
This article incorporates text from this source, which is in the public domain.
- ^ Jackson CB, Hahn D, Schröter B, Richter U, Battersby BJ, Schmitt-Mechelke T, Marttinen P, Nuoffer JM, Schaller A (June 2017). "A novel mitochondrial ATP6 frameshift mutation causing isolated complex V deficiency, ataxia and encephalomyopathy". European Journal of Medical Genetics. 60 (6): 345–351. doi:10.1016/j.ejmg.2017.04.006. hdl:10138/237062. PMID 28412374.
- ^ Imai A, Fujita S, Kishita Y, Kohda M, Tokuzawa Y, Hirata T, Mizuno Y, Harashima H, Nakaya A, Sakata Y, Takeda A, Mori M, Murayama K, Ohtake A, Okazaki Y (March 2016). "Rapidly progressive infantile cardiomyopathy with mitochondrial respiratory chain complex V deficiency due to loss of ATPase 6 and 8 protein". International Journal of Cardiology. 207: 203–5. doi:10.1016/j.ijcard.2016.01.026. PMID 26803244.
Further reading edit
- Torroni A, Achilli A, Macaulay V, Richards M, Bandelt HJ (June 2006). "Harvesting the fruit of the human mtDNA tree". Trends in Genetics. 22 (6): 339–45. doi:10.1016/j.tig.2006.04.001. PMID 16678300.
- Bodenteich A, Mitchell LG, Polymeropoulos MH, Merril CR (May 1992). "Dinucleotide repeat in the human mitochondrial D-loop". Human Molecular Genetics. 1 (2): 140. doi:10.1093/hmg/1.2.140-a. PMID 1301157.
- Lu X, Walker T, MacManus JP, Seligy VL (July 1992). "Differentiation of HT-29 human colonic adenocarcinoma cells correlates with increased expression of mitochondrial RNA: effects of trehalose on cell growth and maturation". Cancer Research. 52 (13): 3718–25. PMID 1377597.
- Marzuki S, Noer AS, Lertrit P, Thyagarajan D, Kapsa R, Utthanaphol P, Byrne E (December 1991). "Normal variants of human mitochondrial DNA and translation products: the building of a reference data base". Human Genetics. 88 (2): 139–45. doi:10.1007/bf00206061. PMID 1757091. S2CID 28048453.
- Moraes CT, Andreetta F, Bonilla E, Shanske S, DiMauro S, Schon EA (March 1991). "Replication-competent human mitochondrial DNA lacking the heavy-strand promoter region". Molecular and Cellular Biology. 11 (3): 1631–7. doi:10.1128/MCB.11.3.1631. PMC 369459. PMID 1996112.
- Attardi G, Chomyn A, Doolittle RF, Mariottini P, Ragan CI (1987). "Seven unidentified reading frames of human mitochondrial DNA encode subunits of the respiratory chain NADH dehydrogenase". Cold Spring Harbor Symposia on Quantitative Biology. 51 Pt 1 (1): 103–14. doi:10.1101/sqb.1986.051.01.013. PMID 3472707.
- Chomyn A, Cleeter MW, Ragan CI, Riley M, Doolittle RF, Attardi G (October 1986). "URF6, last unidentified reading frame of human mtDNA, codes for an NADH dehydrogenase subunit". Science. 234 (4776): 614–8. Bibcode:1986Sci...234..614C. doi:10.1126/science.3764430. PMID 3764430.
- Chomyn A, Mariottini P, Cleeter MW, Ragan CI, Matsuno-Yagi A, Hatefi Y, Doolittle RF, Attardi G (1985). "Six unidentified reading frames of human mitochondrial DNA encode components of the respiratory-chain NADH dehydrogenase". Nature. 314 (6012): 592–7. Bibcode:1985Natur.314..592C. doi:10.1038/314592a0. PMID 3921850. S2CID 32964006.
- Anderson S, Bankier AT, Barrell BG, de Bruijn MH, Coulson AR, Drouin J, Eperon IC, Nierlich DP, Roe BA, Sanger F, Schreier PH, Smith AJ, Staden R, Young IG (April 1981). "Sequence and organization of the human mitochondrial genome". Nature. 290 (5806): 457–65. Bibcode:1981Natur.290..457A. doi:10.1038/290457a0. PMID 7219534. S2CID 4355527.
- Montoya J, Ojala D, Attardi G (April 1981). "Distinctive features of the 5'-terminal sequences of the human mitochondrial mRNAs". Nature. 290 (5806): 465–70. Bibcode:1981Natur.290..465M. doi:10.1038/290465a0. PMID 7219535. S2CID 4358928.
- Horai S, Hayasaka K, Kondo R, Tsugane K, Takahata N (January 1995). "Recent African origin of modern humans revealed by complete sequences of hominoid mitochondrial DNAs". Proceedings of the National Academy of Sciences of the United States of America. 92 (2): 532–6. Bibcode:1995PNAS...92..532H. doi:10.1073/pnas.92.2.532. PMC 42775. PMID 7530363.
- Rieder MJ, Taylor SL, Tobe VO, Nickerson DA (February 1998). "Automating the identification of DNA variations using quality-based fluorescence re-sequencing: analysis of the human mitochondrial genome". Nucleic Acids Research. 26 (4): 967–73. doi:10.1093/nar/26.4.967. PMC 147367. PMID 9461455.
- Andrews RM, Kubacka I, Chinnery PF, Lightowlers RN, Turnbull DM, Howell N (October 1999). "Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA". Nature Genetics. 23 (2): 147. doi:10.1038/13779. PMID 10508508. S2CID 32212178.
- Ingman M, Kaessmann H, Pääbo S, Gyllensten U (December 2000). "Mitochondrial genome variation and the origin of modern humans". Nature. 408 (6813): 708–13. Bibcode:2000Natur.408..708I. doi:10.1038/35047064. PMID 11130070. S2CID 52850476.
- Finnilä S, Lehtonen MS, Majamaa K (June 2001). "Phylogenetic network for European mtDNA". American Journal of Human Genetics. 68 (6): 1475–84. doi:10.1086/320591. PMC 1226134. PMID 11349229.
- Maca-Meyer N, González AM, Larruga JM, Flores C, Cabrera VM (2003). "Major genomic mitochondrial lineages delineate early human expansions". BMC Genetics. 2: 13. doi:10.1186/1471-2156-2-13. PMC 55343. PMID 11553319.
- Herrnstadt C, Elson JL, Fahy E, Preston G, Turnbull DM, Anderson C, Ghosh SS, Olefsky JM, Beal MF, Davis RE, Howell N (May 2002). "Reduced-median-network analysis of complete mitochondrial DNA coding-region sequences for the major African, Asian, and European haplogroups". American Journal of Human Genetics. 70 (5): 1152–71. doi:10.1086/339933. PMC 447592. PMID 11938495.
- Silva WA, Bonatto SL, Holanda AJ, Ribeiro-Dos-Santos AK, Paixão BM, Goldman GH, Abe-Sandes K, Rodriguez-Delfin L, Barbosa M, Paçó-Larson ML, Petzl-Erler ML, Valente V, Santos SE, Zago MA (July 2002). "Mitochondrial genome diversity of Native Americans supports a single early entry of founder populations into America". American Journal of Human Genetics. 71 (1): 187–92. doi:10.1086/341358. PMC 384978. PMID 12022039.
- Mishmar D, Ruiz-Pesini E, Golik P, Macaulay V, Clark AG, Hosseini S, Brandon M, Easley K, Chen E, Brown MD, Sukernik RI, Olckers A, Wallace DC (January 2003). "Natural selection shaped regional mtDNA variation in humans". Proceedings of the National Academy of Sciences of the United States of America. 100 (1): 171–6. Bibcode:2003PNAS..100..171M. doi:10.1073/pnas.0136972100. PMC 140917. PMID 12509511.
- Ingman M, Gyllensten U (July 2003). "Mitochondrial genome variation and evolutionary history of Australian and New Guinean aborigines". Genome Research. 13 (7): 1600–6. doi:10.1101/gr.686603. PMC 403733. PMID 12840039.
This article incorporates text from the United States National Library of Medicine, which is in the public domain.