


Summary
In organic chemistry the Brook rearrangement refers to any [1,n] carbon to oxygen silyl migration. The rearrangement was first observed in the late 1950s by Canadian chemist Adrian Gibbs Brook (1924–2013), after which the reaction is named.[1] These migrations can be promoted in a number of different ways, including thermally, photolytically or under basic/acidic conditions.[2] In the forward direction, these silyl migrations produce silyl ethers as products which is driven by the stability of the oxygen-silicon bond.

The silyl substituents can be aliphatic or aromatic, and if the silicon is a center of chirality, the migration occurs with retention at this center. This migration occurs through a transition state where silicon is penta-coordinate and bears a partial negative charge.[3] If a center of chirality is present at the carbon center to which the silyl group is attached, then inversion occurs at this center. As an example, if (trimethylsilyl)methanol where to be deprotonated, a [1,2]-Brook rearrangement would occur.
Reaction mechanism edit
The reaction mechanism for this rearrangement depends on the conditions employed to affect the rearrangement and the nature of the starting material.[2] Anionic rearrangements are the most common Brook rearrangements observed, and their mechanisms can be broken into two general categories. The first category starts with proton abstraction of a nearby hydroxyl group by a base. This generates an alkoxide which then acts as a nucleophile and attacks the silicon atom in a nucleophilic displacement reaction, with the methylene group acting as the leaving group. The generated carbanion is then protonated by the H-B species to form the product. In the case where the base used is consumed in the reaction (i.e. Butyllithium), then the carbanion can act as a base to deprotonate further starting material to generate the final product.
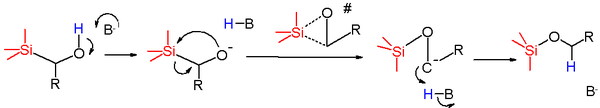
The proposed transition state for this reaction step is a three-membered ring, with significant negative charge build-up on the carbon atom and the silicon atom, as demonstrated by Hammett sigma and rho studies. This reaction generally proceeds with a low activation energy and a large negative entropy of activation. This further supports the cyclic three member transition state, as this would be considerably more ordered than the ground state of the starting material. The reaction proceeds with overall retention at the silicon center, as demonstrated with a Walden Cycle (shown below). This supports a pentacoordinate silicon as part of the mechanism, as trigonal bipyramidal geometry around the silicon with one of the O or C axial and the other equatorial would explain the observed retention in configuration at the silicon center. This mechanism also proceeds with inversion at the carbon center.
This reaction is known to be reversible. Depending on the relative stabilities of the carbanion and oxy-anion formed, a silyl ether is perfectly capable of rearranging to a species with the silicon bonded to the carbon atom, and the free alcohol being present. This would be termed a Retro-Brook rearrangement.
The second category of anionic brook rearrangements involves nucleophilic attack at an sp2 hybridized center to generate an oxy-anion two atoms removed from the silicon atom. This can then undergoes intramolecular attack by the oxy-anion to yield the silyl ether, but the final fate of the carbanion often depends on the substrate in question. For example, attempting to perform a Wittig reaction on acylsilane results in the formation of a silyl enol ether instead of the expected alkene, due to elimination by the carbanion instead of protonation as seen above.

The Brook rearrangement has been shown to occur with retention of configuration at the silicon center as demonstrated in the following Walden cycle:

All steps in this cycle were known to proceed with retention of configuration except for attack of the lithium reagent (which proceeded by inversion) and the Brook Rearrangement, which was being investigated. By starting with a chiral silicon of known configuration, the stereochemistry of the reaction could be determined by looking at the specific rotation of the recovered silane. Since it is known that attack by the lithium reagent proceeds with inversion, the recovered silane should be the opposite enantiomer of the starting silane (single inversion) if the Brook Rearrangement proceeds with retention, and the same enantiomer if the reaction proceeds with inversion (double inversion). Experimentally, the recovered silane was the opposite enantiomer, showing that the reaction occurred with retention at the silicon center.
Scope edit
Brook rearrangements are known in acylsilanes.[4] Beyond that, acylsilanes are well known for their hydrolysis in basic solution to a silanol and an aldehyde. This occurs through a Brook-rearrangement initiated by attack at the carbonyl group. A related reaction, involving initial attack at the silicon center, causes migration of one of the silicon groups to the carbonyl carbon, which initiates a Brook-Rearrangement. If the silicon group was chiral, the end product is a chiral silyl ether, as the migration occurs stereospecifically.
Rearrangements analogous to the Brook Rearrangement are known for many other types of atoms. These include nitrogen, phosphorus,[5] and sulfur as the nucleophilic component, with boron and germanium analogous known as the electrophilic component.
References edit
- ^ Brook, A. G. (1958). "Isomerism of some α-hydroxysilanes to silyl ethers". J. Am. Chem. Soc. 80 (8): 1886–1889. doi:10.1021/ja01541a026.
- ^ a b Brook, A. G.; Bassindale, A. R. (1980). "Chapter 9. Molecular rearrangements of organosilicon compounds". Rearrangements in Ground and Excited States, Vol 2. New York: Academic Press. pp. 149–221. ISBN 9781483218724.
- ^ Brook, A. G. (1974). "Molecular rearrangements of organosilicon compounds". Acc. Chem. Res. 7 (3): 77–84. doi:10.1021/ar50075a003.
- ^ Patrocinio, Amauri F. and Moran, Paulo J. S. Acylsilanes and their applications in organic chemistry. J. Braz. Chem. Soc., 2001, vol.12, no.1, p.07-31. ISSN 0103-5053. Online article
- ^ Engel, Robert; Cohen, JamieLee Iolani (2004). Synthesis of Carbon-Phosphorus Bonds (2nd ed.). CRC Press. ISBN 0-8493-1617-0. LCCN 2003060796.
{{cite book}}: CS1 maint: multiple names: authors list (link)