


Summary
Actinium is a chemical element; it has symbol Ac and atomic number 89. It was first isolated by Friedrich Oskar Giesel in 1902, who gave it the name emanium; the element got its name by being wrongly identified with a substance André-Louis Debierne found in 1899 and called actinium. Actinium gave the name to the actinide series, a set of 15 elements between actinium and lawrencium in the periodic table. Together with polonium, radium, and radon, actinium was one of the first non-primordial radioactive elements to be isolated.
 | ||||||||||||||||||||||||||||||||||
| Actinium | ||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pronunciation | /ækˈtɪniəm/ | |||||||||||||||||||||||||||||||||
| Appearance | silvery-white, glowing with an eerie blue light;[1] sometimes with a golden cast[2] | |||||||||||||||||||||||||||||||||
| Mass number | [227] | |||||||||||||||||||||||||||||||||
| Actinium in the periodic table | ||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||
| Atomic number (Z) | 89 | |||||||||||||||||||||||||||||||||
| Group | f-block groups (no number) | |||||||||||||||||||||||||||||||||
| Period | period 7 | |||||||||||||||||||||||||||||||||
| Block | f-block | |||||||||||||||||||||||||||||||||
| Electron configuration | [Rn] 6d1 7s2 | |||||||||||||||||||||||||||||||||
| Electrons per shell | 2, 8, 18, 32, 18, 9, 2 | |||||||||||||||||||||||||||||||||
| Physical properties | ||||||||||||||||||||||||||||||||||
| Phase at STP | solid | |||||||||||||||||||||||||||||||||
| Melting point | 1500 K (1227 °C, 2240 °F) (estimated)[2] | |||||||||||||||||||||||||||||||||
| Boiling point | 3500±300 K (3200±300 °C, 5800±500 °F) (extrapolated)[2] | |||||||||||||||||||||||||||||||||
| Density (near r.t.) | 10 g/cm3 | |||||||||||||||||||||||||||||||||
| Heat of fusion | 14 kJ/mol | |||||||||||||||||||||||||||||||||
| Heat of vaporization | 400 kJ/mol | |||||||||||||||||||||||||||||||||
| Molar heat capacity | 27.2 J/(mol·K) | |||||||||||||||||||||||||||||||||
| Atomic properties | ||||||||||||||||||||||||||||||||||
| Oxidation states | +3 (a strongly basic oxide) | |||||||||||||||||||||||||||||||||
| Electronegativity | Pauling scale: 1.1 | |||||||||||||||||||||||||||||||||
| Ionization energies |
| |||||||||||||||||||||||||||||||||
| Covalent radius | 215 pm | |||||||||||||||||||||||||||||||||
 | ||||||||||||||||||||||||||||||||||
| Other properties | ||||||||||||||||||||||||||||||||||
| Natural occurrence | from decay | |||||||||||||||||||||||||||||||||
| Crystal structure | face-centered cubic (fcc) | |||||||||||||||||||||||||||||||||
| Thermal conductivity | 12 W/(m⋅K) | |||||||||||||||||||||||||||||||||
| CAS Number | 7440-34-8 | |||||||||||||||||||||||||||||||||
| History | ||||||||||||||||||||||||||||||||||
| Discovery and first isolation | Friedrich Oskar Giesel (1902, 1903) | |||||||||||||||||||||||||||||||||
| Named by | André-Louis Debierne (1899) | |||||||||||||||||||||||||||||||||
| Isotopes of actinium | ||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||
A soft, silvery-white radioactive metal, actinium reacts rapidly with oxygen and moisture in air forming a white coating of actinium oxide that prevents further oxidation. As with most lanthanides and many actinides, actinium assumes oxidation state +3 in nearly all its chemical compounds. Actinium is found only in traces in uranium and thorium ores as the isotope 227Ac, which decays with a half-life of 21.772 years, predominantly emitting beta and sometimes alpha particles, and 228Ac, which is beta active with a half-life of 6.15 hours. One tonne of natural uranium in ore contains about 0.2 milligrams of actinium-227, and one tonne of thorium contains about 5 nanograms of actinium-228. The close similarity of physical and chemical properties of actinium and lanthanum makes separation of actinium from the ore impractical. Instead, the element is prepared, in milligram amounts, by the neutron irradiation of 226Ra in a nuclear reactor. Owing to its scarcity, high price and radioactivity, actinium has no significant industrial use. Its current applications include a neutron source and an agent for radiation therapy.
History edit
André-Louis Debierne, a French chemist, announced the discovery of a new element in 1899. He separated it from pitchblende residues left by Marie and Pierre Curie after they had extracted radium. In 1899, Debierne described the substance as similar to titanium[4] and (in 1900) as similar to thorium.[5] Friedrich Oskar Giesel found in 1902[6] a substance similar to lanthanum and called it "emanium" in 1904.[7] After a comparison of the substances' half-lives determined by Debierne,[8] Harriet Brooks in 1904, and Otto Hahn and Otto Sackur in 1905, Debierne's chosen name for the new element was retained because it had seniority, despite the contradicting chemical properties he claimed for the element at different times.[9][10]
Articles published in the 1970s[11] and later[12] suggest that Debierne's results published in 1904 conflict with those reported in 1899 and 1900. Furthermore, the now-known chemistry of actinium precludes its presence as anything other than a minor constituent of Debierne's 1899 and 1900 results; in fact, the chemical properties he reported make it likely that he had, instead, accidentally identified protactinium, which would not be discovered for another fourteen years, only to have it disappear due to its hydrolysis and adsorption onto his laboratory equipment. This has led some authors to advocate that Giesel alone should be credited with the discovery.[2] A less confrontational vision of scientific discovery is proposed by Adloff.[12] He suggests that hindsight criticism of the early publications should be mitigated by the then nascent state of radiochemistry: highlighting the prudence of Debierne's claims in the original papers, he notes that nobody can contend that Debierne's substance did not contain actinium.[12] Debierne, who is now considered by the vast majority of historians as the discoverer, lost interest in the element and left the topic. Giesel, on the other hand, can rightfully be credited with the first preparation of radiochemically pure actinium and with the identification of its atomic number 89.[11]
The name actinium originates from the Ancient Greek aktis, aktinos (ακτίς, ακτίνος), meaning beam or ray.[13] Its symbol Ac is also used in abbreviations of other compounds that have nothing to do with actinium, such as acetyl, acetate[14] and sometimes acetaldehyde.[15]
Properties edit
Actinium is a soft, silvery-white,[16][17] radioactive, metallic element. Its estimated shear modulus is similar to that of lead.[18] Owing to its strong radioactivity, actinium glows in the dark with a pale blue light, which originates from the surrounding air ionized by the emitted energetic particles.[19] Actinium has similar chemical properties to lanthanum and other lanthanides, and therefore these elements are difficult to separate when extracting from uranium ores. Solvent extraction and ion chromatography are commonly used for the separation.[20]
The first element of the actinides, actinium gave the set its name, much as lanthanum had done for the lanthanides. The actinides are much more diverse than the lanthanides[21] and therefore it was not until 1945 that the most significant change to Dmitri Mendeleev's periodic table since the recognition of the lanthanides, the introduction of the actinides, was generally accepted after Glenn T. Seaborg's research on the transuranium elements[22] (although it had been proposed as early as 1892 by British chemist Henry Bassett).[23]
Actinium reacts rapidly with oxygen and moisture in air forming a white coating of actinium oxide that impedes further oxidation.[16] As with most lanthanides and actinides, actinium exists in the oxidation state +3, and the Ac3+ ions are colorless in solutions.[24] The oxidation state +3 originates from the [Rn] 6d17s2 electronic configuration of actinium, with three valence electrons that are easily donated to give the stable closed-shell structure of the noble gas radon.[17] Although the 5f orbitals are unoccupied in an actinium atom, it can be used as a valence orbital in actinium complexes and hence it is generally considered the first 5f element by authors working on it.[25][26][27] Ac3+ is the largest of all known tripositive ions and its first coordination sphere contains approximately 10.9 ± 0.5 water molecules.[28]
Chemical compounds edit
Due to actinium's intense radioactivity, only a limited number of actinium compounds are known. These include: AcF3, AcCl3, AcBr3, AcOF, AcOCl, AcOBr, Ac2S3, Ac2O3, AcPO4 and Ac(NO3)3. They all contain actinium in the oxidation state +3.[24][29] In particular, the lattice constants of the analogous lanthanum and actinium compounds differ by only a few percent.[29]
| Formula | color | symmetry | space group | No | Pearson symbol | a (pm) | b (pm) | c (pm) | Z | density, g/cm3 |
|---|---|---|---|---|---|---|---|---|---|---|
| Ac | silvery | fcc[30] | Fm3m | 225 | cF4 | 531.1 | 531.1 | 531.1 | 4 | 10.07 |
| AcH2 | unknown | cubic[30] | Fm3m | 225 | cF12 | 567 | 567 | 567 | 4 | 8.35 |
| Ac2O3 | white[16] | trigonal[31] | P3m1 | 164 | hP5 | 408 | 408 | 630 | 1 | 9.18 |
| Ac2S3 | black | cubic[32] | I43d | 220 | cI28 | 778.56 | 778.56 | 778.56 | 4 | 6.71 |
| AcF3 | white[33] | hexagonal[29][31] | P3c1 | 165 | hP24 | 741 | 741 | 755 | 6 | 7.88 |
| AcCl3 | white | hexagonal[29][34] | P63/m | 165 | hP8 | 764 | 764 | 456 | 2 | 4.8 |
| AcBr3 | white[29] | hexagonal[34] | P63/m | 165 | hP8 | 764 | 764 | 456 | 2 | 5.85 |
| AcOF | white[35] | cubic[29] | Fm3m | 593.1 | 8.28 | |||||
| AcOCl | white | tetragonal[29] | 424 | 424 | 707 | 7.23 | ||||
| AcOBr | white | tetragonal[29] | 427 | 427 | 740 | 7.89 | ||||
| AcPO4·0.5H2O | unknown | hexagonal[29] | 721 | 721 | 664 | 5.48 |
Here a, b and c are lattice constants, No is space group number and Z is the number of formula units per unit cell. Density was not measured directly but calculated from the lattice parameters.
Oxides edit
Actinium oxide (Ac2O3) can be obtained by heating the hydroxide at 500 °C or the oxalate at 1100 °C, in vacuum. Its crystal lattice is isotypic with the oxides of most trivalent rare-earth metals.[29]
Halides edit
Actinium trifluoride can be produced either in solution or in solid reaction. The former reaction is carried out at room temperature, by adding hydrofluoric acid to a solution containing actinium ions. In the latter method, actinium metal is treated with hydrogen fluoride vapors at 700 °C in an all-platinum setup. Treating actinium trifluoride with ammonium hydroxide at 900–1000 °C yields oxyfluoride AcOF. Whereas lanthanum oxyfluoride can be easily obtained by burning lanthanum trifluoride in air at 800 °C for an hour, similar treatment of actinium trifluoride yields no AcOF and only results in melting of the initial product.[29][35]
- AcF3 + 2 NH3 + H2O → AcOF + 2 NH4F
Actinium trichloride is obtained by reacting actinium hydroxide or oxalate with carbon tetrachloride vapors at temperatures above 960 °C. Similar to oxyfluoride, actinium oxychloride can be prepared by hydrolyzing actinium trichloride with ammonium hydroxide at 1000 °C. However, in contrast to the oxyfluoride, the oxychloride could well be synthesized by igniting a solution of actinium trichloride in hydrochloric acid with ammonia.[29]
Reaction of aluminium bromide and actinium oxide yields actinium tribromide:
- Ac2O3 + 2 AlBr3 → 2 AcBr3 + Al2O3
and treating it with ammonium hydroxide at 500 °C results in the oxybromide AcOBr.[29]
Other compounds edit
Actinium hydride was obtained by reduction of actinium trichloride with potassium at 300 °C, and its structure was deduced by analogy with the corresponding LaH2 hydride. The source of hydrogen in the reaction was uncertain.[36]
Mixing monosodium phosphate (NaH2PO4) with a solution of actinium in hydrochloric acid yields white-colored actinium phosphate hemihydrate (AcPO4·0.5H2O), and heating actinium oxalate with hydrogen sulfide vapors at 1400 °C for a few minutes results in a black actinium sulfide Ac2S3. It may possibly be produced by acting with a mixture of hydrogen sulfide and carbon disulfide on actinium oxide at 1000 °C.[29]
Isotopes edit
Naturally occurring actinium is principally composed of two radioactive isotopes; 227
Ac (from the radioactive family of 235
U) and 228
Ac (a granddaughter of 232
Th). 227
Ac decays mainly as a beta emitter with a very small energy, but in 1.38% of cases it emits an alpha particle, so it can readily be identified through alpha spectrometry.[2] Thirty-three radioisotopes have been identified, the most stable being 227
Ac with a half-life of 21.772 years, 225
Ac with a half-life of 10.0 days and 226
Ac with a half-life of 29.37 hours. All remaining radioactive isotopes have half-lives that are less than 10 hours and the majority of them have half-lives shorter than one minute. The shortest-lived known isotope of actinium is 217
Ac (half-life of 69 nanoseconds) which decays through alpha decay. Actinium also has two known meta states.[37] The most significant isotopes for chemistry are 225Ac, 227Ac, and 228Ac.[2]
Purified 227
Ac comes into equilibrium with its decay products after about a half of year. It decays according to its 21.772-year half-life emitting mostly beta (98.62%) and some alpha particles (1.38%);[37] the successive decay products are part of the actinium series. Owing to the low available amounts, low energy of its beta particles (maximum 44.8 keV) and low intensity of alpha radiation, 227
Ac is difficult to detect directly by its emission and it is therefore traced via its decay products.[24] The isotopes of actinium range in atomic weight from 203 u (203
Ac) to 236 u (236
Ac).[37]
| Isotope | Production | Decay | Half-life |
|---|---|---|---|
| 221Ac | 232Th(d,9n)→225Pa(α)→221Ac | α | 52 ms |
| 222Ac | 232Th(d,8n)→226Pa(α)→222Ac | α | 5.0 s |
| 223Ac | 232Th(d,7n)→227Pa(α)→223Ac | α | 2.1 min |
| 224Ac | 232Th(d,6n)→228Pa(α)→224Ac | α | 2.78 hours |
| 225Ac | 232Th(n,γ)→233Th(β−)→233Pa(β−)→233U(α)→229Th(α)→225Ra(β−)→225Ac | α | 10 days |
| 226Ac | 226Ra(d,2n)→226Ac | α, β− electron capture |
29.37 hours |
| 227Ac | 235U(α)→231Th(β−)→231Pa(α)→227Ac | α, β− | 21.77 years |
| 228Ac | 232Th(α)→228Ra(β−)→228Ac | β− | 6.15 hours |
| 229Ac | 228Ra(n,γ)→229Ra(β−)→229Ac | β− | 62.7 min |
| 230Ac | 232Th(d,α)→230Ac | β− | 122 s |
| 231Ac | 232Th(γ,p)→231Ac | β− | 7.5 min |
| 232Ac | 232Th(n,p)→232Ac | β− | 119 s |
Occurrence and synthesis edit

Actinium is found only in traces in uranium ores – one tonne of uranium in ore contains about 0.2 milligrams of 227Ac[38][39] – and in thorium ores, which contain about 5 nanograms of 228Ac per one tonne of thorium. The actinium isotope 227Ac is a transient member of the uranium-actinium series decay chain, which begins with the parent isotope 235U (or 239Pu) and ends with the stable lead isotope 207Pb. The isotope 228Ac is a transient member of the thorium series decay chain, which begins with the parent isotope 232Th and ends with the stable lead isotope 208Pb. Another actinium isotope (225Ac) is transiently present in the neptunium series decay chain, beginning with 237Np (or 233U) and ending with thallium (205Tl) and near-stable bismuth (209Bi); even though all primordial 237Np has decayed away, it is continuously produced by neutron knock-out reactions on natural 238U.
The low natural concentration, and the close similarity of physical and chemical properties to those of lanthanum and other lanthanides, which are always abundant in actinium-bearing ores, render separation of actinium from the ore impractical. The most concentrated actinium sample prepared from raw material consisted of 7 micrograms of 227Ac in less than 0.1 milligrams of La2O3, and complete separation was never achieved.[40] Instead, actinium is prepared, in milligram amounts, by the neutron irradiation of 226Ra in a nuclear reactor.[39][41]
![{\displaystyle {\ce {^{226}_{88}Ra + ^{1}_{0}n -> ^{227}_{88}Ra ->[\beta^-][42.2 \ {\ce {min}}] ^{227}_{89}Ac}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/0971e4ce21fbf7bb4673856bff635b1a64d11fb2)
The reaction yield is about 2% of the radium weight. 227Ac can further capture neutrons resulting in small amounts of 228Ac. After the synthesis, actinium is separated from radium and from the products of decay and nuclear fusion, such as thorium, polonium, lead and bismuth. The extraction can be performed with thenoyltrifluoroacetone-benzene solution from an aqueous solution of the radiation products, and the selectivity to a certain element is achieved by adjusting the pH (to about 6.0 for actinium).[38] An alternative procedure is anion exchange with an appropriate resin in nitric acid, which can result in a separation factor of 1,000,000 for radium and actinium vs. thorium in a two-stage process. Actinium can then be separated from radium, with a ratio of about 100, using a low cross-linking cation exchange resin and nitric acid as eluant.[42]
225Ac was first produced artificially at the Institute for Transuranium Elements (ITU) in Germany using a cyclotron and at St George Hospital in Sydney using a linac in 2000.[43] This rare isotope has potential applications in radiation therapy and is most efficiently produced by bombarding a radium-226 target with 20–30 MeV deuterium ions. This reaction also yields 226Ac which however decays with a half-life of 29 hours and thus does not contaminate 225Ac.[44]
Actinium metal has been prepared by the reduction of actinium fluoride with lithium vapor in vacuum at a temperature between 1100 and 1300 °C. Higher temperatures resulted in evaporation of the product and lower ones lead to an incomplete transformation. Lithium was chosen among other alkali metals because its fluoride is most volatile.[13][16]
Applications edit
Owing to its scarcity, high price and radioactivity, 227Ac currently has no significant industrial use, but 225Ac is currently being studied for use in cancer treatments such as targeted alpha therapies.[13][27] 227Ac is highly radioactive and was therefore studied for use as an active element of radioisotope thermoelectric generators, for example in spacecraft. The oxide of 227Ac pressed with beryllium is also an efficient neutron source with the activity exceeding that of the standard americium-beryllium and radium-beryllium pairs.[45] In all those applications, 227Ac (a beta source) is merely a progenitor which generates alpha-emitting isotopes upon its decay. Beryllium captures alpha particles and emits neutrons owing to its large cross-section for the (α,n) nuclear reaction:

The 227AcBe neutron sources can be applied in a neutron probe – a standard device for measuring the quantity of water present in soil, as well as moisture/density for quality control in highway construction.[46][47] Such probes are also used in well logging applications, in neutron radiography, tomography and other radiochemical investigations.[48]
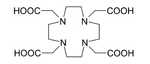
225Ac is applied in medicine to produce 213Bi in a reusable generator[42] or can be used alone as an agent for radiation therapy, in particular targeted alpha therapy (TAT). This isotope has a half-life of 10 days, making it much more suitable for radiation therapy than 213Bi (half-life 46 minutes).[27] Additionally, 225Ac decays to nontoxic 209Bi rather than toxic lead, which is the final product in the decay chains of several other candidate isotopes, namely 227Th, 228Th, and 230U.[27] Not only 225Ac itself, but also its daughters, emit alpha particles which kill cancer cells in the body. The major difficulty with application of 225Ac was that intravenous injection of simple actinium complexes resulted in their accumulation in the bones and liver for a period of tens of years. As a result, after the cancer cells were quickly killed by alpha particles from 225Ac, the radiation from the actinium and its daughters might induce new mutations. To solve this problem, 225Ac was bound to a chelating agent, such as citrate, ethylenediaminetetraacetic acid (EDTA) or diethylene triamine pentaacetic acid (DTPA). This reduced actinium accumulation in the bones, but the excretion from the body remained slow. Much better results were obtained with such chelating agents as HEHA (1,4,7,10,13,16-hexaazacyclohexadecane-N,N′,N″,N‴,N‴′,N‴″-hexaacetic acid)[49] or DOTA (1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid) coupled to trastuzumab, a monoclonal antibody that interferes with the HER2/neu receptor. The latter delivery combination was tested on mice and proved to be effective against leukemia, lymphoma, breast, ovarian, neuroblastoma and prostate cancers.[50][51][52]
The medium half-life of 227Ac (21.77 years) makes it a very convenient radioactive isotope in modeling the slow vertical mixing of oceanic waters. The associated processes cannot be studied with the required accuracy by direct measurements of current velocities (of the order 50 meters per year). However, evaluation of the concentration depth-profiles for different isotopes allows estimating the mixing rates. The physics behind this method is as follows: oceanic waters contain homogeneously dispersed 235U. Its decay product, 231Pa, gradually precipitates to the bottom, so that its concentration first increases with depth and then stays nearly constant. 231Pa decays to 227Ac; however, the concentration of the latter isotope does not follow the 231Pa depth profile, but instead increases toward the sea bottom. This occurs because of the mixing processes which raise some additional 227Ac from the sea bottom. Thus analysis of both 231Pa and 227Ac depth profiles allows researchers to model the mixing behavior.[53][54]
There are theoretical predictions that AcHx hydrides (in this case with very high pressure) are a candidate for a near room-temperature superconductor as they have Tc significantly higher than H3S, possibly near 250 K.[55]
Precautions edit
227Ac is highly radioactive and experiments with it are carried out in a specially designed laboratory equipped with a tight glove box. When actinium trichloride is administered intravenously to rats, about 33% of actinium is deposited into the bones and 50% into the liver. Its toxicity is comparable to, but slightly lower than that of americium and plutonium.[56] For trace quantities, fume hoods with good aeration suffice; for gram amounts, hot cells with shielding from the intense gamma radiation emitted by 227Ac are necessary.[57]
See also edit
Notes edit
References edit
- ^ Wall, Greg (8 September 2003). "C&EN: It's Elemental: The Periodic Table - Actinium". C&EN: It's Elemental: The Periodic Table. Chemical and Engineering News. Retrieved 2 June 2011.
- ^ a b c d e f Kirby, Harold W.; Morss, Lester R. (2006). "Actinium". The Chemistry of the Actinide and Transactinide Elements. p. 18. doi:10.1007/1-4020-3598-5_2. ISBN 978-1-4020-3555-5.
- ^ Kondev, F. G.; Wang, M.; Huang, W. J.; Naimi, S.; Audi, G. (2021). "The NUBASE2020 evaluation of nuclear properties" (PDF). Chinese Physics C. 45 (3): 030001. doi:10.1088/1674-1137/abddae.
- ^ Debierne, André-Louis (1899). "Sur un nouvelle matière radio-active". Comptes Rendus (in French). 129: 593–595.
- ^ Debierne, André-Louis (1900–1901). "Sur un nouvelle matière radio-actif – l'actinium". Comptes Rendus (in French). 130: 906–908.
- ^ Giesel, Friedrich Oskar (1902). "Ueber Radium und radioactive Stoffe". Berichte der Deutschen Chemischen Gesellschaft (in German). 35 (3): 3608–3611. doi:10.1002/cber.190203503187.
- ^ Giesel, Friedrich Oskar (1904). "Ueber den Emanationskörper (Emanium)". Berichte der Deutschen Chemischen Gesellschaft (in German). 37 (2): 1696–1699. doi:10.1002/cber.19040370280.
- ^ Debierne, André-Louis (1904). "Sur l'actinium". Comptes Rendus (in French). 139: 538–540.
- ^ Giesel, Friedrich Oskar (1904). "Ueber Emanium". Berichte der Deutschen Chemischen Gesellschaft (in German). 37 (2): 1696–1699. doi:10.1002/cber.19040370280.
- ^ Giesel, Friedrich Oskar (1905). "Ueber Emanium". Berichte der Deutschen Chemischen Gesellschaft (in German). 38 (1): 775–778. doi:10.1002/cber.190503801130.
- ^ a b Kirby, Harold W. (1971). "The Discovery of Actinium". Isis. 62 (3): 290–308. doi:10.1086/350760. JSTOR 229943. S2CID 144651011.
- ^ a b c Adloff, J. P. (2000). "The centenary of a controversial discovery: actinium". Radiochim. Acta. 88 (3–4_2000): 123–128. doi:10.1524/ract.2000.88.3-4.123. S2CID 94016074.
- ^ a b c Hammond, C. R. The Elements in Lide, D. R., ed. (2005). CRC Handbook of Chemistry and Physics (86th ed.). Boca Raton (FL): CRC Press. ISBN 0-8493-0486-5.
- ^ Gilley, Cynthia Brooke; University of California, San Diego. Chemistry (2008). New convertible isocyanides for the Ugi reaction; application to the stereoselective synthesis of omuralide. p. 11. ISBN 978-0-549-79554-4.
- ^ Reimers, Jeffrey R. (2011). Computational Methods for Large Systems: Electronic Structure Approaches for Biotechnology and Nanotechnology. John Wiley and Sons. p. 575. ISBN 978-0-470-48788-4.
- ^ a b c d Stites, Joseph G.; Salutsky, Murrell L.; Stone, Bob D. (1955). "Preparation of Actinium Metal". J. Am. Chem. Soc. 77 (1): 237–240. doi:10.1021/ja01606a085.
- ^ a b Actinium, in Encyclopædia Britannica, 15th edition, 1995, p. 70
- ^ Seitz, Frederick and Turnbull, David (1964) Solid state physics: advances in research and applications. Academic Press. ISBN 0-12-607716-9 pp. 289–291
- ^ Richard A. Muller (2010). Physics and Technology for Future Presidents: An Introduction to the Essential Physics Every World Leader Needs to Know. Princeton University Press. pp. 136–. ISBN 978-0-691-13504-5.
- ^ Katz, J. J.; Manning, W. M. (1952). "Chemistry of the Actinide Elements". Annual Review of Nuclear Science. 1: 245–262. Bibcode:1952ARNPS...1..245K. doi:10.1146/annurev.ns.01.120152.001333.
- ^ Jørgensen, Christian (1973). "The Loose Connection between Electron Configuration and the Chemical Behavior of the Heavy Elements (Transuranics)". Angewandte Chemie International Edition. 12 (1): 12–19. doi:10.1002/anie.197300121.
- ^ Seaborg, Glenn T. (1946). "The Transuranium Elements". Science. 104 (2704): 379–386. Bibcode:1946Sci...104..379S. doi:10.1126/science.104.2704.379. JSTOR 1675046. PMID 17842184.
- ^ Thyssen, P.; Binnemans, K. (2011). Gschneidner, K. A. Jr.; Bünzli, J-C.G; Vecharsky, Bünzli (eds.). Accommodation of the Rare Earths in the Periodic Table: A Historical Analysis. Vol. 41. Amsterdam: Elsevier. pp. 1–94. doi:10.1016/B978-0-444-53590-0.00001-7. ISBN 978-0-444-53590-0.
{{cite book}}:|journal=ignored (help) - ^ a b c Actinium, Great Soviet Encyclopedia (in Russian)
- ^ Tomeček, Josef; Li, Cen; Schreckenbach, Georg (2023). "Actinium coordination chemistry: A density functional theory study with monodentate and bidentate ligands". Journal of Computational Chemistry. 44 (3): 334–345. doi:10.1002/jcc.26929. PMID 35668552. S2CID 249433367.
- ^ Deblonde, Gauthier J.-P.; Zavarin, Mavrik; Kersting, Annie B. (2021). "The coordination properties and ionic radius of actinium: A 120-year-old enigma". Coordination Chemistry Reviews. 446: 214130. doi:10.1016/j.ccr.2021.214130.
- ^ a b c d Deblonde, Gauthier J.-P.; Abergel, Rebecca J. (21 October 2016). "Active actinium". Nature Chemistry. 8 (11): 1084. Bibcode:2016NatCh...8.1084D. doi:10.1038/nchem.2653. ISSN 1755-4349. OSTI 1458479. PMID 27768109.
- ^ Ferrier, Maryline G.; Stein, Benjamin W.; Batista, Enrique R.; Berg, John M.; Birnbaum, Eva R.; Engle, Jonathan W.; John, Kevin D.; Kozimor, Stosh A.; Lezama Pacheco, Juan S.; Redman, Lindsay N. (2017). "Synthesis and Characterization of the Actinium Aquo Ion". ACS Central Science. 3 (3): 176–185. doi:10.1021/acscentsci.6b00356. PMC 5364452. PMID 28386595.
- ^ a b c d e f g h i j k l m n Fried, Sherman; Hagemann, French; Zachariasen, W. H. (1950). "The Preparation and Identification of Some Pure Actinium Compounds". Journal of the American Chemical Society. 72 (2): 771–775. doi:10.1021/ja01158a034.
- ^ a b Farr, J.; Giorgi, A. L.; Bowman, M. G.; Money, R. K. (1961). "The crystal structure of actinium metal and actinium hydride". Journal of Inorganic and Nuclear Chemistry. 18: 42–47. doi:10.1016/0022-1902(61)80369-2. OSTI 4397640.
- ^ a b Zachariasen, W. H. (1949). "Crystal chemical studies of the 5f-series of elements. XII. New compounds representing known structure types". Acta Crystallographica. 2 (6): 388–390. doi:10.1107/S0365110X49001016.
- ^ Zachariasen, W. H. (1949). "Crystal chemical studies of the 5f-series of elements. VI. The Ce2S3-Ce3S4 type of structure" (PDF). Acta Crystallographica. 2: 57–60. doi:10.1107/S0365110X49000126. Archived (PDF) from the original on 9 October 2022.
- ^ Meyer, p. 71
- ^ a b Zachariasen, W. H. (1948). "Crystal chemical studies of the 5f-series of elements. I. New structure types". Acta Crystallographica. 1 (5): 265–268. doi:10.1107/S0365110X48000703.
- ^ a b Meyer, pp. 87–88
- ^ Meyer, p. 43
- ^ a b c Audi, G.; Kondev, F. G.; Wang, M.; Huang, W. J.; Naimi, S. (2017). "The NUBASE2016 evaluation of nuclear properties" (PDF). Chinese Physics C. 41 (3): 030001. Bibcode:2017ChPhC..41c0001A. doi:10.1088/1674-1137/41/3/030001.
- ^ a b Hagemann, French (1950). "The Isolation of Actinium". Journal of the American Chemical Society. 72 (2): 768–771. doi:10.1021/ja01158a033.
- ^ a b Greenwood, Norman N.; Earnshaw, Alan (1997). Chemistry of the Elements (2nd ed.). Butterworth-Heinemann. p. 946. ISBN 978-0-08-037941-8.
- ^ Kirby, H. W.; Morss, Lester R. (2010). Morss, Lester R.; Edelstein, Norman M.; Fuger, Jean (eds.). Actinium. Dordrecht: Springer Netherlands. pp. 18–51. doi:10.1007/978-94-007-0211-0_2. ISBN 978-94-007-0210-3.
- ^ Emeleus, H. J. (1987). Advances in inorganic chemistry and radiochemistry. Academic Press. pp. 16–. ISBN 978-0-12-023631-2.
- ^ a b Bolla, Rose A.; Malkemus, D.; Mirzadeh, S. (2005). "Production of actinium-225 for alpha particle mediated radioimmunotherapy". Applied Radiation and Isotopes. 62 (5): 667–679. doi:10.1016/j.apradiso.2004.12.003. PMID 15763472.
- ^ Melville, G; Allen, Bj (2009). "Cyclotron and linac production of Ac-225". Applied Radiation and Isotopes. 67 (4): 549–55. doi:10.1016/j.apradiso.2008.11.012. PMID 19135381.
- ^ Russell, Pamela J.; Jackson, Paul and Kingsley, Elizabeth Anne (2003) Prostate cancer methods and protocols[permanent dead link]. Humana Press. ISBN 0-89603-978-1, p. 336
- ^ Russell, Alan M. and Lee, Kok Loong (2005) Structure-property relations in nonferrous metals. Wiley. ISBN 0-471-64952-X, pp. 470–471
- ^ Majumdar, D. K. (2004) Irrigation Water Management: Principles and Practice. ISBN 81-203-1729-7 p. 108
- ^ Chandrasekharan, H. and Gupta, Navindu (2006) Fundamentals of Nuclear Science – Application in Agriculture. ISBN 81-7211-200-9 pp. 202 ff
- ^ Dixon, W. R.; Bielesch, Alice; Geiger, K. W. (1957). "Neutron Spectrum of an Actinium–Beryllium Source". Can. J. Phys. 35 (6): 699–702. Bibcode:1957CaJPh..35..699D. doi:10.1139/p57-075.
- ^ Deal K.A.; Davis I.A.; Mirzadeh S.; Kennel S.J. & Brechbiel M.W. (1999). "Improved in Vivo Stability of Actinium-225 Macrocyclic Complexes". J Med Chem. 42 (15): 2988–9. doi:10.1021/jm990141f. PMID 10425108.
- ^ McDevitt, Michael R.; Ma, Dangshe; Lai, Lawrence T.; et al. (2001). "Tumor Therapy with Targeted Atomic Nanogenerators". Science. 294 (5546): 1537–1540. Bibcode:2001Sci...294.1537M. doi:10.1126/science.1064126. PMID 11711678. S2CID 11782419.
- ^ Borchardt, Paul E.; et al. (2003). "Targeted Actinium-225 in Vivo Generators for Therapy of Ovarian Cancer" (PDF). Cancer Research. 63 (16): 5084–5090. PMID 12941838. Archived (PDF) from the original on 9 October 2022.
- ^ Ballangrud, A. M.; et al. (2004). "Alpha-particle emitting atomic generator (Actinium-225)-labeled trastuzumab (herceptin) targeting of breast cancer spheroids: efficacy versus HER2/neu expression". Clinical Cancer Research. 10 (13): 4489–97. doi:10.1158/1078-0432.CCR-03-0800. PMID 15240541.
- ^ Nozaki, Yoshiyuki (1984). "Excess 227Ac in deep ocean water". Nature. 310 (5977): 486–488. Bibcode:1984Natur.310..486N. doi:10.1038/310486a0. S2CID 4344946.
- ^ Geibert, W.; Rutgers Van Der Loeff, M. M.; Hanfland, C.; Dauelsberg, H.-J. (2002). "Actinium-227 as a deep-sea tracer: sources, distribution and applications". Earth and Planetary Science Letters. 198 (1–2): 147–165. Bibcode:2002E&PSL.198..147G. doi:10.1016/S0012-821X(02)00512-5.
- ^ Semenok, Dmitrii V.; Kvashnin, Alexander G.; Kruglov, Ivan A.; Oganov, Artem R. (19 April 2018). "Actinium hydrides AcH10, AcH12, AcH16 as high-temperature conventional superconductors". The Journal of Physical Chemistry Letters. 9 (8): 1920–1926. arXiv:1802.05676. doi:10.1021/acs.jpclett.8b00615. ISSN 1948-7185. PMID 29589444. S2CID 4620593.
- ^ Langham, W.; Storer, J. (1952). "Toxicology of Actinium Equilibrium Mixture". Los Alamos Scientific Lab.: Technical Report. doi:10.2172/4406766.
- ^ Keller, Cornelius; Wolf, Walter; Shani, Jashovam. "Radionuclides, 2. Radioactive Elements and Artificial Radionuclides". Ullmann's Encyclopedia of Industrial Chemistry. Weinheim: Wiley-VCH. doi:10.1002/14356007.o22_o15. ISBN 978-3527306732.
Bibliography edit
- Meyer, Gerd and Morss, Lester R. (1991) Synthesis of lanthanide and actinide compounds, Springer. ISBN 0-7923-1018-7
External links edit
- Actinium at The Periodic Table of Videos (University of Nottingham)
- NLM Hazardous Substances Databank – Actinium, Radioactive
- Actinium in Kirby, H. W.; Morss, L. R. (2006). Morss; Edelstein, Norman M.; Fuger, Jean (eds.). The Chemistry of the Actinide and Transactinide Elements (3rd ed.). Dordrecht, The Netherlands: Springer. ISBN 978-1-4020-3555-5.