


Summary
In physical chemistry, the Arrhenius equation is a formula for the temperature dependence of reaction rates. The equation was proposed by Svante Arrhenius in 1889, based on the work of Dutch chemist Jacobus Henricus van 't Hoff who had noted in 1884 that the van 't Hoff equation for the temperature dependence of equilibrium constants suggests such a formula for the rates of both forward and reverse reactions. This equation has a vast and important application in determining the rate of chemical reactions and for calculation of energy of activation. Arrhenius provided a physical justification and interpretation for the formula.[1][2][3][4] Currently, it is best seen as an empirical relationship.[5]: 188 It can be used to model the temperature variation of diffusion coefficients, population of crystal vacancies, creep rates, and many other thermally induced processes and reactions. The Eyring equation, developed in 1935, also expresses the relationship between rate and energy.
Equation edit




The Arrhenius equation gives the dependence of the rate constant of a chemical reaction on the absolute temperature as

- k is the rate constant (frequency of collisions resulting in a reaction),
- T is the absolute temperature,
- A is the pre-exponential factor or Arrhenius factor or frequency factor. Arrhenius originally considered A to be a temperature-independent constant for each chemical reaction.[6] However more recent treatments include some temperature dependence – see § Modified Arrhenius equation below.
- Ea is the molar activation energy for the reaction,
- R is the universal gas constant.[1][2][4]
Alternatively, the equation may be expressed as

- Ea is the activation energy for the reaction (in the same unit as kBT),
- kB is the Boltzmann constant.
The only difference is the unit of Ea: the former form uses energy per mole, which is common in chemistry, while the latter form uses energy per molecule directly, which is common in physics. The different units are accounted for in using either the gas constant, R, or the Boltzmann constant, kB, as the multiplier of temperature T.
The unit of the pre-exponential factor A are identical to those of the rate constant and will vary depending on the order of the reaction. If the reaction is first order it has the unit s−1, and for that reason it is often called the frequency factor or attempt frequency of the reaction. Most simply, k is the number of collisions that result in a reaction per second, A is the number of collisions (leading to a reaction or not) per second occurring with the proper orientation to react[7] and is the probability that any given collision will result in a reaction. It can be seen that either increasing the temperature or decreasing the activation energy (for example through the use of catalysts) will result in an increase in rate of reaction.

Given the small temperature range of kinetic studies, it is reasonable to approximate the activation energy as being independent of the temperature. Similarly, under a wide range of practical conditions, the weak temperature dependence of the pre-exponential factor is negligible compared to the temperature dependence of the factor ; except in the case of "barrierless" diffusion-limited reactions, in which case the pre-exponential factor is dominant and is directly observable.
With this equation it can be roughly estimated that the rate of reaction increases by a factor of about 2 to 3 for every 10 °C rise in temperature, for common values of activation energy and temperature range.[8]
The factor denotes the fraction of molecules with energy greater than or equal to .[9]


Derivation edit
Van't Hoff argued that the temperature of a reaction and the standard equilibrium constant exhibit the relation:


-
(1)

where denotes the apposite standard internal energy change value.

Let and respectively denote the forward and backward reaction rates of the reaction of interest, then ,[10] an equation from which naturally follows.




Substituting the expression for in eq.(1), we obtain .


The preceding equation can be broken down into the following two equations:
-
(2)

and
-
(3)

where and are the activation energies associated with the forward and backward reactions respectively, with .



Experimental findings suggest that the constants in eq.(2) and eq.(3) can be treated as being equal to zero, so that
-
()

and
-
()

Integrating these equations and taking the exponential yields the results and , where each pre-exponential factor or is mathematically the exponential of the constant of integration for the respective indefinite integral in question.[11]




Arrhenius plot edit
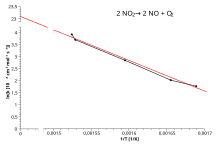
Taking the natural logarithm of Arrhenius equation yields:

Rearranging yields:

This has the same form as an equation for a straight line:

So, when a reaction has a rate constant that obeys the Arrhenius equation, a plot of ln k versus T−1 gives a straight line, whose gradient and intercept can be used to determine Ea and A. This procedure has become so common in experimental chemical kinetics that practitioners have taken to using it to define the activation energy for a reaction. That is, the activation energy is defined to be (−R) times the slope of a plot of ln k vs. (1/T):
![{\displaystyle E_{\text{a}}\equiv -R\left[{\frac {\partial \ln k}{\partial (1/T)}}\right]_{P}.}](https://wikimedia.org/api/rest_v1/media/math/render/svg/1001eea5bd180a6d3b04793c72490514b41d80fa)
Modified Arrhenius equation edit
The modified Arrhenius equation[12] makes explicit the temperature dependence of the pre-exponential factor. The modified equation is usually of the form

The original Arrhenius expression above corresponds to n = 0. Fitted rate constants typically lie in the range −1 < n < 1. Theoretical analyses yield various predictions for n. It has been pointed out that "it is not feasible to establish, on the basis of temperature studies of the rate constant, whether the predicted T1/2 dependence of the pre-exponential factor is observed experimentally".[5]: 190 However, if additional evidence is available, from theory and/or from experiment (such as density dependence), there is no obstacle to incisive tests of the Arrhenius law.
Another common modification is the stretched exponential form[citation needed]
![{\displaystyle k=A\exp \left[-\left({\frac {E_{a}}{RT}}\right)^{\beta }\right],}](https://wikimedia.org/api/rest_v1/media/math/render/svg/89847c364b7f6ebd0bbd35c26fb4e0d66b5fd679)
Theoretical interpretation of the equation edit
Arrhenius's concept of activation energy edit
Arrhenius argued that for reactants to transform into products, they must first acquire a minimum amount of energy, called the activation energy Ea. At an absolute temperature T, the fraction of molecules that have a kinetic energy greater than Ea can be calculated from statistical mechanics. The concept of activation energy explains the exponential nature of the relationship, and in one way or another, it is present in all kinetic theories.
The calculations for reaction rate constants involve an energy averaging over a Maxwell–Boltzmann distribution with as lower bound and so are often of the type of incomplete gamma functions, which turn out to be proportional to .

Collision theory edit
One approach is the collision theory of chemical reactions, developed by Max Trautz and William Lewis in the years 1916–18. In this theory, molecules are supposed to react if they collide with a relative kinetic energy along their line of centers that exceeds Ea. The number of binary collisions between two unlike molecules per second per unit volume is found to be[13]

The rate constant is then calculated as , so that the collision theory predicts that the pre-exponential factor is equal to the collision number zAB. However for many reactions this agrees poorly with experiment, so the rate constant is written instead as . Here is an empirical steric factor, often much less than 1.00, which is interpreted as the fraction of sufficiently energetic collisions in which the two molecules have the correct mutual orientation to react.[13]



Transition state theory edit
The Eyring equation, another Arrhenius-like expression, appears in the "transition state theory" of chemical reactions, formulated by Eugene Wigner, Henry Eyring, Michael Polanyi and M. G. Evans in the 1930s. The Eyring equation can be written:






At first sight this looks like an exponential multiplied by a factor that is linear in temperature. However, free energy is itself a temperature dependent quantity. The free energy of activation is the difference of an enthalpy term and an entropy term multiplied by the absolute temperature. The pre-exponential factor depends primarily on the entropy of activation. The overall expression again takes the form of an Arrhenius exponential (of enthalpy rather than energy) multiplied by a slowly varying function of T. The precise form of the temperature dependence depends upon the reaction, and can be calculated using formulas from statistical mechanics involving the partition functions of the reactants and of the activated complex.

Limitations of the idea of Arrhenius activation energy edit
Both the Arrhenius activation energy and the rate constant k are experimentally determined, and represent macroscopic reaction-specific parameters that are not simply related to threshold energies and the success of individual collisions at the molecular level. Consider a particular collision (an elementary reaction) between molecules A and B. The collision angle, the relative translational energy, the internal (particularly vibrational) energy will all determine the chance that the collision will produce a product molecule AB. Macroscopic measurements of E and k are the result of many individual collisions with differing collision parameters. To probe reaction rates at molecular level, experiments are conducted under near-collisional conditions and this subject is often called molecular reaction dynamics.[15]
Another situation where the explanation of the Arrhenius equation parameters falls short is in heterogeneous catalysis, especially for reactions that show Langmuir-Hinshelwood kinetics. Clearly, molecules on surfaces do not "collide" directly, and a simple molecular cross-section does not apply here. Instead, the pre-exponential factor reflects the travel across the surface towards the active site.[16]
There are deviations from the Arrhenius law during the glass transition in all classes of glass-forming matter.[17] The Arrhenius law predicts that the motion of the structural units (atoms, molecules, ions, etc.) should slow down at a slower rate through the glass transition than is experimentally observed. In other words, the structural units slow down at a faster rate than is predicted by the Arrhenius law. This observation is made reasonable assuming that the units must overcome an energy barrier by means of a thermal activation energy. The thermal energy must be high enough to allow for translational motion of the units which leads to viscous flow of the material.
See also edit
- Accelerated aging
- Eyring equation
- Q10 (temperature coefficient)
- Van 't Hoff equation
- Clausius–Clapeyron relation
- Gibbs–Helmholtz equation
- Cherry blossom front – predicted using the Arrhenius equation
References edit
- ^ a b Arrhenius, S. A. (1889). "Über die Dissociationswärme und den Einfluß der Temperatur auf den Dissociationsgrad der Elektrolyte". Z. Phys. Chem. 4: 96–116. doi:10.1515/zpch-1889-0408. S2CID 202553486.
- ^ a b Arrhenius, S. A. (1889). "Über die Reaktionsgeschwindigkeit bei der Inversion von Rohrzucker durch Säuren". Z. Phys. Chem. 4: 226–48. doi:10.1515/zpch-1889-0416. S2CID 100032801.
- ^ Laidler, K. J. (1984). "The development of the Arrhenius equation". J. Chem. Educ. 61 (6): 494–498. Bibcode:1984JChEd..61..494L. doi:10.1021/ed061p494.
- ^ a b Laidler, K. J. (1987) Chemical Kinetics, Third Edition, Harper & Row, p. 42
- ^ a b Kenneth Connors, Chemical Kinetics, 1990, VCH Publishers Chemical Kinetics: The Study of Reaction Rates in Solution at Google Books
- ^ IUPAC Goldbook definition of Arrhenius equation.
- ^ Silberberg, Martin S. (2006). Chemistry (fourth ed.). NY: McGraw-Hill. p. 696. ISBN 0-07-111658-3.
- ^ Avery, H. E. (1974). "4. Dependence of Rate on Temperature". Basic Reaction Kinetics and Mechanisms. Springer. pp. 47–58. Retrieved 18 December 2023.
However, the rate of reaction varies greatly with temperature, since for a typical process the rate doubles or trebles for a rise in temperature of 10 °C.
- ^ "6.2.3.3: The Arrhenius Law – Activation Energies". Chemistry LibreTexts. 2013-10-02.
- ^ "15.2: The Equilibrium Constant (K)". Chemistry LibreTexts. 2016-03-11. Retrieved 2023-06-27.
- ^ "Arrhenius Equation" (PDF). University of Pennsylvania. Retrieved 27 June 2023.
- ^ IUPAC Goldbook definition of modified Arrhenius equation.
- ^ a b Laidler, Keith J.; Meiser, John H. (1982). Physical Chemistry (1st ed.). Benjamin/Cummings. pp. 376–78. ISBN 0-8053-5682-7.
- ^ Laidler, Keith J.; Meiser, John H. (1982). Physical Chemistry (1st ed.). Benjamin/Cummings. pp. 378–83. ISBN 0-8053-5682-7.
- ^ Levine, R.D. (2005) Molecular Reaction Dynamics, Cambridge University Press
- ^ Slot, Thierry K.; Riley, Nathan; Shiju, N. Raveendran; Medlin, J. Will; Rothenberg, Gadi (2020). "An experimental approach for controlling confinement effects at catalyst interfaces". Chemical Science. 11 (40): 11024–11029. doi:10.1039/D0SC04118A. ISSN 2041-6520. PMC 8162257. PMID 34123192.
- ^ Bauer, Th.; Lunkenheimer, P.; Loidl, A. (2013). "Cooperativity and the Freezing of Molecular Motion at the Glass Transition". Physical Review Letters. 111 (22): 225702. arXiv:1306.4630. Bibcode:2013PhRvL.111v5702B. doi:10.1103/PhysRevLett.111.225702. PMID 24329455. S2CID 13720989.
Bibliography edit
- Pauling, L. C. (1988). General Chemistry. Dover Publications.
- Laidler, K. J. (1987). Chemical Kinetics (3rd ed.). Harper & Row.
- Laidler, K. J. (1993). The World of Physical Chemistry. Oxford University Press.
External links edit
- Carbon Dioxide solubility in Polyethylene – Using Arrhenius equation for calculating species solubility in polymers