


Summary
A persistent carbene (also known as stable carbene) is an organic molecule whose natural resonance structure has a carbon atom with incomplete octet (a carbene), but does not exhibit the tremendous instability typically associated with such moieties. The best-known examples and by far largest subgroup are the N-heterocyclic carbenes (NHC)[1] (sometimes called Arduengo carbenes), in which nitrogen atoms flank the formal carbene.

Modern theoretical analysis suggests that the term "persistent carbene" is in fact a misnomer. Persistent carbenes do not in fact have a carbene electronic structure in their ground state, but instead an ylide stabilized by aromatic resonance or steric shielding. Excitation to a carbene structure then accounts for the carbene-like dimerization that some persistent carbenes undergo over the course of days.
Persistent carbenes in general, and Arduengo carbenes in particular, are popular ligands in organometallic chemistry.
History edit
Early evidence edit
In 1957, Ronald Breslow proposed that a relatively stable nucleophilic carbene, a thiazol-2-ylidene derivative of vitamin B1 (thiamine), was the catalyst involved in the benzoin condensation that yields furoin from furfural.[2] [3] In this cycle, the vitamin's thiazolium ring exchanges a hydrogen atom (attached to carbon 2 of the ring) for a furfural residue. In deuterated water, the C2-proton was found to rapidly exchange for a deuteron in a statistical equilibrium.[4]
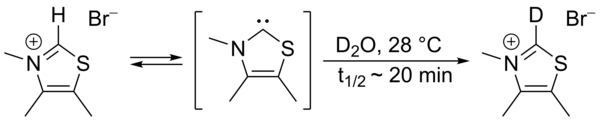
This exchange was proposed to proceed via intermediacy of a thiazol-2-ylidene. In 2012 the isolation of the so-called Breslow intermediate was reported.[5][6]
In 1960, Hans-Werner Wanzlick and coworkers conjectured that carbenes derived from dihydroimidazol-2-ylidene were produced by vacuum pyrolysis of the corresponding 2-trichloromethyl dihydroimidazole compounds with the loss of chloroform.[7][8] [9] They conjectured that the carbene existed in equilibrium with its dimer, a tetraaminoethylene derivative, the so-called Wanzlick equilibrium. This conjecture was challenged by Lemal and coworkers in 1964, who presented evidence that the dimer did not dissociate;[10] and by Winberg in 1965.[11] However, subsequent experiments by Denk, Herrmann and others have confirmed this equilibrium, albeit in specific circumstances.[12][13]
Isolation of persistent carbenes edit
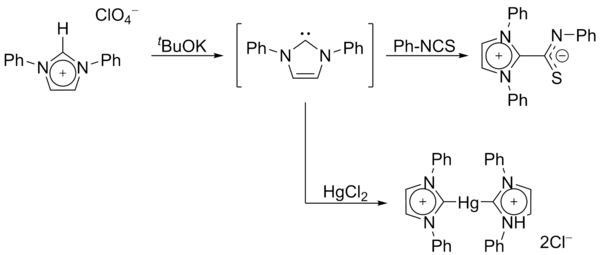
In 1970, Wanzlick's group generated imidazol-2-ylidene carbenes by the deprotonation of an imidazolium salt.[14] Wanzlick as well as Roald Hoffmann,[9][15] proposed that these imidazole-based carbenes should be more stable than their 4,5-dihydro analogues, due to Hückel-type aromaticity. Wanzlick did not however isolate imidazol-2-ylidenes, but instead their coordination compounds with mercury and isothiocyanate:
In 1988, Guy Bertrand and others isolated a phosphinocarbene. These species can be represented as either a λ3-phosphinocarbene or λ5-phosphaacetylene:[16][17]

These compounds were called "push-pull carbenes" in reference to the contrasting electron affinities of the phosphorus and silicon atoms. They exhibit both carbenic and alkynic reactivity. An X-ray structure of this molecule has not been obtained and at the time of publication some doubt remained as to their exact carbenic nature.
In 1991, Arduengo and coworkers crystallized a diaminocarbene by deprotonation of an imidazolium cation:[18]

This carbene, the forerunner of a large family of carbenes with the imidazol-2-ylidene core, is indefinitely stable at room temperature in the absence of oxygen and moisture. It melts at 240–241 °C without decomposition. The 13C NMR spectrum shows a signal at 211 ppm for the carbenic atom.[19] The X-ray structure revealed longer N–C bond lengths in the ring of the carbene than in the parent imidazolium compound, indicating that there was very little double bond character to these bonds.[20]
The first air-stable ylidic carbene, a chlorinated member of the imidazol-2-ylidene family, was obtained in 1997.[21]
In 2000, Bertrand obtained additional carbenes of the phosphanyl type, including (phosphanyl)(trifluoromethyl)carbene, stable in solution at -30 °C[22] and a moderately stable (amino)(aryl)carbene with only one heteroatom adjacent to the carbenic atom.[23][24]
Stabilization through adjacent orbitals edit

In the modern understanding, the superficially unoccupied p-orbital on a (meta)stable carbene is not, in fact, fully empty. Instead, the carbene Lewis structures are in resonance with dative bonds toward adjacent lone-pair or pi-bond orbitals.[25]

Early workers attributed the stability of Arduengo carbenes to the bulky N-adamantyl substituents, which prevent the carbene from dimerising. But replacement of the N-adamantyl groups with methyl groups also affords 1,3,4,5-tetramethylimidazol-2‑ylidene (Me4ImC:), a thermodynamically stable unhindered NHC.[26]

In 1995, Arduengo's group obtained a carbene derivative of dihydroimidazol-2-ylidene, proving that stability did not arise from the aromaticity of the conjugated imidazole backbone.[27] The following year, the first acyclic persistent carbene demonstrated that stability did not even require a cyclic backbone.[28] Unhindered derivatives of the hydrogenated[29][30] and acyclic[30][31][32] carbenes dimerized, suggesting that Me4ImC: might be exceptional, rather than paradigmatic. But the behavior of the acyclic carbenes offered a tantalizing clue to the stabilization mechanism. [citation needed]
Unlike the cyclic derivatives, acyclic carbenes are flexible and bonds to the carbenic atom admit rotation. But bond rotation in the compound appeared hindered, suggesting a double bond character that would place the positive charge on adjacent nitrogen atoms while preserving the octet rule.[28] Indeed, most persistent carbenes are stabilized by two flanking nitrogen centers. The outliers include an aminothiocarbene and an aminooxycarbene, which use other heteroatoms,[33][34] and room-temperature-stable bis(diisopropylamino)cyclopropenylidene, in which the carbene atom is connected to two carbon atoms in a three-member, aromatic, cyclopropenylidene ring.[35]

Classes of stable carbenes edit
The following are examples of the classes of stable carbenes isolated to date:
Imidazol-2-ylidenes edit
The first stable carbenes to be isolated were based on an imidazole ring, with the hydrogen in carbon 2 of the ring (between the two nitrogen atoms) removed, and other hydrogens replaced by various groups. These imidazol-2-ylidenes are still the most stable and the most well studied and understood family of persistent carbenes.[citation needed]
A considerable range of imidazol-2-ylidenes have been synthesised, including those in which the 1,3-positions have been functionalised with alkyl, aryl,[26] alkyloxy, alkylamino, alkylphosphino[36] and even chiral substituents:[36]


(View the 3D structure with external viewer.)
In particular, substitution of two chlorine atoms for the two hydrogens at ring positions 4 and 5 yielded the first air-stable carbene.[21] Its extra stability probably results from the electron-withdrawing effect of the chlorine substituents, which reduce the electron density on the carbon atom bearing the lone pair, via induction through the sigma-backbone.
Molecules containing two and even three imidazol-2-ylidene groups have also been synthesised.[37][38]
Imidazole-based carbenes are thermodynamically stable and generally have diagnostic 13C NMR chemical shift values between 210 and 230 ppm for the carbenic carbon. Typically, X-ray structures of these molecules show N–C–N bond angles of 101–102°.[citation needed]
Triazol-5-ylidenes edit
Depending on the arrangement of the three nitrogen atoms in triazol-5-ylidene, there are two possible isomers, namely 1,2,3-triazol-5-ylidenes and 1,2,4-triazol-5-ylidenes.

The triazol-5-ylidenes based on the 1,2,4-triazole ring are pictured below and were first prepared by Enders and coworkers[39] by vacuum pyrolysis through loss of methanol from 2-methoxytriazoles. Only a limited range of these molecules have been reported, with the triphenyl substituted molecule being commercially available.

Triazole-based carbenes are thermodynamically stable and have diagnostic 13C NMR chemical shift values between 210 and 220 ppm for the carbenic carbon. The X-ray structure of the triphenyl substituted carbene above shows an N–C–N bond angle of around 101°. The 5-methoxytriazole precursor to this carbene was made by the treatment of a triazolium salt with sodium methoxide, which attacks as a nucleophile.[39] This may indicate that these carbenes are less aromatic than imidazol-2-ylidenes, as the imidazolium precursors do not react with nucleophiles due to the resultant loss of aromaticity.[citation needed]
Other diaminocarbenes edit
The two families above can be seen as special cases of a broader class of compounds which have a carbenic atom bridging two nitrogen atoms. A range of such diaminocarbenes have been prepared principally by Roger Alder's research group. In some of these compounds, the N–C–N unit is a member of a five- or six-membered non-aromatic ring,[27][29][40] including a bicyclic example. In other examples, the adjacent nitrogens are connected only through the carbenic atom, and may or may not be part of separate rings.[28][31][32]

Unlike the aromatic imidazol-2-ylidenes or triazol-5-ylidenes, these carbenes appear not to be thermodynamically stable, as shown by the dimerisation of some unhindered cyclic and acyclic examples.[29][31] Studies[30] suggest that these carbenes dimerise via acid catalysed dimerisation (as in the Wanzlick equilibrium).
Diaminocarbenes have diagnostic 13C NMR chemical shift values between 230 and 270 ppm for the carbenic atom. The X-ray structure of dihydroimidazole-2-ylidene shows a N–C–N bond angle of about 106°, whilst the angle of the acyclic carbene is 121°, both greater than those seen for imidazol-2-ylidenes.
Heteroamino carbenes edit
There exist several variants of the stable carbenes above where one of the nitrogen atoms adjacent to the carbene center (the α nitrogens) has been replaced by an alternative heteroatom, such as oxygen, sulfur, or phosphorus.[16][17][33][34]

In particular, the formal substitution of sulfur for one of the nitrogens in imidazole would yield the aromatic heterocyclic compound thiazole. A thiazole based carbene (analogous to the carbene postulated by Breslow)[41] has been prepared and characterised by X-ray crystallography.[33] Other non-aromatic aminocarbenes with O, S and P atoms adjacent (i.e. alpha) to the carbene centre have been prepared, for example, thio- and oxyiminium based carbenes have been characterised by X-ray crystallography.[34]
Since oxygen and sulfur are divalent, steric protection of the carbenic centre is limited especially when the N–C–X unit is part of a ring. These acyclic carbenes have diagnostic 13C NMR chemical shift values between 250 and 300 ppm for the carbenic carbon, further downfield than any other types of stable carbene. X-ray structures have shown N–C–X bond angles of around 104° and 109° respectively.[citation needed]
Carbenes that formally derive from imidazole-2-ylidenes by substitution of sulfur, oxygen, or other chalcogens for both α-nitrogens are expected to be unstable, as they have the potential to dissociate into an alkyne (R1C≡CR2) and a carbon dichalcogenide (X1=C=X2).[42][43]
Non-amino carbenes edit
The reaction of carbon disulfide (CS2) with electron deficient acetylene derivatives is proposed to give transient 1,3-dithiolium carbenes (i.e. where X1 = X2 = S), which then dimerise to give derivatives of tetrathiafulvene. Thus it is possible that the reverse of this process might be occurring in similar carbenes.[42][43]
Bertrand's carbenes edit
In Bertrand's persistent carbenes, the unsaturated carbon is bonded to a phosphorus and a silicon.[44] However, these compounds seem to exhibit some alkynic properties, and when published the exact carbenic nature of these red oils was in debate.[17]
Other nucleophilic carbenes edit
One stable N-heterocyclic carbene[45] has a structure analogous to borazine with one boron atom replaced by a methylene group. This results in a planar six-electron compound.

Cyclopropenylidenes edit
Another family of carbenes is based on a cyclopropenylidene core, a three-carbon ring with a double bond between the two atoms adjacent to the carbenic one. This family is exemplified by bis(diisopropylamino)cyclopropenylidene.[35]
Triplet state carbenes edit
Persistent carbenes tend to exist in the singlet, dimerizing when forced into triplet states. Nevertheless, Hideo Tomioka and associates used electron delocalization to produce a comparatively stable triplet carbene (bis(9-anthryl)carbene) in 2001. It has an unusually long half-life of 19 minutes.[46][47]
Although the figure below shows the two parts of the molecule in one flat plane, molecular geometry puts the two aromatic parts in orthogonal positions with respect to each other.

In 2006 a triplet carbene was reported by the same group with a half-life of 40 minutes.[48] This carbene is prepared by a photochemical decomposition of a diazomethane precursor by 300 nm light in benzene with expulsion of nitrogen gas.
Again the figure below is not an adequate representation of the actual molecular structure: both phenyl rings are positioned orthogonal with respect to each other. The carbene carbon has an sp-hybridisation, the two remaining orthogonal p-orbitals each conjugating with one of the aromatic rings.

Exposure to oxygen (a triplet diradical) converts this carbene to the corresponding benzophenone. The diphenylmethane compound is formed when it is trapped by cyclohexa-1,4-diene. As with the other carbenes, this species contains large bulky substituents, namely bromine and the trifluoromethyl groups on the phenyl rings, that shield the carbene and prevent or slow down the process of dimerization to a 1,1,2,2-tetra(phenyl)alkene. Based on computer simulations, the distance of the divalent carbon atom to its neighbors is claimed to be 138 picometers with a bond angle of 158.8°. The planes of the phenyl groups are almost at right angles to each other (the dihedral angle being 85.7°).
Mesoionic carbenes edit
Mesoionic carbenes (MICs) are similar to N-heterocyclic carbenes (NHCs) except that canonical resonance structures with the carbene depicted cannot be drawn without adding additional charges. Mesoionic carbenes are also referred to as abnormal N-heterocyclic carbenes (aNHC) or remote N-heterocyclic carbenes (rNHC). A variety of free carbenes can be isolated and are stable at room temperature. Other free carbenes are not stable and are susceptible to intermolecular decomposition pathways.[citation needed]
Chemical properties edit
Basicity and nucleophilicity edit
The imidazol-2-ylidenes are strong bases, having pKa ≈ 24 for the conjugate acid in dimethyl sulfoxide (DMSO):[49]

However, further work showed that diaminocarbenes will deprotonate the DMSO solvent, with the resulting anion reacting with the resulting amidinium salt.

Reaction of imidazol-2-ylidenes with 1-bromohexane gave 90% of the 2-substituted adduct, with only 10% of the corresponding alkene, indicating that these molecules are also reasonably nucleophilic.
pKa values for the conjugate acids of several NHC families have been examined in aqueous solution. pKa values of triazolium ions lie in the range 16.5–17.8,[50] around 3 pKa units more acidic than related imidazolium ions.[51]
Dimerisation edit
At one time, stable carbenes were thought to reversibly dimerise through the so-called Wanzlick equilibrium. However, imidazol-2-ylidenes and triazol-5-ylidenes are thermodynamically stable and do not dimerise, and have been stored in solution in the absence of water and air for years. This is presumably due to the aromatic nature of these carbenes, which is lost upon dimerisation. In fact imidazol-2-ylidenes are so thermodynamically stable that only in highly constrained conditions are these carbenes forced to dimerise.
Chen and Taton[52] made a doubly tethered diimidazol-2-ylidene by deprotonating the respective diimidazolium salt. Only the deprotonation of the doubly tethered diimidazolium salt with the shorter methylene bridge (–CH2–) resulted in the dicarbene dimer:

If this dimer existed as a dicarbene, the electron lone pairs on the carbenic carbon would be forced into close proximity. Presumably the resulting repulsive electrostatic interactions would have a significant destabilising effect. To avoid this electronic interaction, the carbene units dimerise.
On the other hand, heteroamino carbenes (such as R2N–C–OR or R2N–C–SR) and non-aromatic carbenes such as diaminocarbenes (such as R2N–C–NR2) have been shown to dimerise,[53] albeit quite slowly. This has been presumed to be due to the high barrier to singlet state dimerisation:

Diaminocarbenes do not truly dimerise, but rather form the dimer by reaction via formamidinium salts, a protonated precursor species.[30] Accordingly, this reaction can be acid catalysed. This reaction occurs because unlike imidazolium based carbenes, there is no loss of aromaticity in protonation of the carbene.
Unlike the dimerisation of triplet state carbenes, these singlet state carbenes do not approach head to head ("least motion"), but rather the carbene lone pair attacks the empty carbon p-orbital ("non-least motion"). Carbene dimerisation can be catalyzed by both acids and metals.
Reactivity edit
The chemistry of stable carbenes has not been fully explored. However, Enders et al.[39][54][55] have performed a range of organic reactions involving a triazol-5-ylidene. These reactions are outlined below and may be considered as a model for other carbenes.

| a | 3,6-diphenyl-1,2,4,5-tetrazine, toluene | 92% | e | 2 equiv., PhNCO, toluene, reflux | 92% | |
|---|---|---|---|---|---|---|
| b | RXH, RT | 95–97% | f | CS2, toluene, or PhNCS, THF, RT | 71–90% | |
| c | O2, S8, or Se, toluene, reflux | 54–68% | g | Maleimide, THF, RT | 47–84% | |
| d | R1CH=CHR2, THF, RT | 25–68% | h | Dimethylacetylene dicarboxylate, THF, reflux | 21% |
These carbenes tend to behave in a nucleophilic fashion (e and f), performing insertion reactions (b), addition reactions (c), [2+1] cycloadditions (d, g and h), [4+1] cycloadditions (a) as well as simple deprotonations. The insertion reactions (b) probably proceed via deprotonation, resulting in the generation of a nucleophile (−XR) which can attack the generated salt giving the impression of a H–X insertion.
The reported stable isothiazole carbene (2b) derived from an isothiazolium perchlorate (1)[56] was questioned.[57] The researchers were only able to isolate 2-imino-2H-thiete (4). The intermediate 3 was proposed through a rearrangement reaction. The carbene 2b is no longer considered as stable.[58]

Carbene complexation edit
Imidazol-2-ylidenes, triazol-5-ylidenes (and less so, diaminocarbenes) have been shown to coordinate to a plethora of elements, from alkali metals, main group elements, transition metals and even lanthanides and actinides. A periodic table of elements gives some idea of the complexes which have been prepared, and in many cases these have been identified by single crystal X-ray crystallography.[40][59][60] Stable carbenes are believed to behave in a similar fashion to organophosphines in their coordination properties to metals. These ligands are said to be good σ-donors through the carbenic lone pair, but poor π-acceptors due to internal ligand back-donation from the nitrogen atoms adjacent to the carbene centre, and so are able to coordinate to even relatively electron deficient metals. Enders [61] and Hermann[62][63] have shown that these carbenes are suitable replacements for phosphine ligands in several catalytic cycles. Whilst they have found that these ligands do not activate the metal catalyst as much as phosphine ligands they often result in more robust catalysts. Several catalytic systems have been looked into by Hermann and Enders, using catalysts containing imidazole and triazole carbene ligands, with moderate success.[59][61][62][63] Grubbs [64] has reported replacing a phosphine ligand (PCy3) with an imidazol-2-ylidene in the olefin metathesis catalyst RuCl2(PCy3)2CHPh, and noted increased ring closing metathesis as well as exhibiting "a remarkable air and water stability". Molecules containing two and three carbene moieties have been prepared as potential bidentate and tridentate carbene ligands.[37][38]
| Group → | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ↓ Period | ||||||||||||||||||||
| 1 | 1 H |
2 He | ||||||||||||||||||
| 2 | 3 Li |
4 Be |
5 B |
6 C |
7 N |
8 O |
9 F |
10 Ne | ||||||||||||
| 3 | 11 Na |
12 Mg |
13 Al |
14 Si |
15 P |
16 S |
17 Cl |
18 Ar | ||||||||||||
| 4 | 19 K |
20 Ca |
21 Sc |
22 Ti |
23 V |
24 Cr |
25 Mn |
26 Fe |
27 Co |
28 Ni |
29 Cu |
30 Zn |
31 Ga |
32 Ge |
33 As |
34 Se |
35 Br |
36 Kr | ||
| 5 | 37 Rb |
38 Sr |
39 Y |
40 Zr |
41 Nb |
42 Mo |
43 Tc |
44 Ru |
45 Rh |
46 Pd |
47 Ag |
48 Cd |
49 In |
50 Sn |
51 Sb |
52 Te |
53 I |
54 Xe | ||
| 6 | 55 Cs |
56 Ba |
71 Lu |
72 Hf |
73 Ta |
74 W |
75 Re |
76 Os |
77 Ir |
78 Pt |
79 Au |
80 Hg |
81 Tl |
82 Pb |
83 Bi |
84 Po |
85 At |
86 Rn | ||
| 7 | 87 Fr |
88 Ra |
103 Lr |
104 Rf |
105 Db |
106 Sg |
107 Bh |
108 Hs |
109 Mt |
110 Ds |
111 Rg |
112 Cn |
113 Nh |
114 Fl |
115 Mc |
116 Lv |
117 Ts |
118 Og | ||
| 57 La |
58 Ce |
59 Pr |
60 Nd |
61 Pm |
62 Sm |
63 Eu |
64 Gd |
65 Tb |
66 Dy |
67 Ho |
68 Er |
69 Tm |
70 Yb | |||||||
| 89 Ac |
90 Th |
91 Pa |
92 U |
93 Np |
94 Pu |
95 Am |
96 Cm |
97 Bk |
98 Cf |
99 Es |
100 Fm |
101 Md |
102 No | |||||||
- Legend
- Carbene complex with element known
- No carbene complex with element known
Carbenes in organometallic chemistry & catalysis edit
Carbenes can be stabilised as organometallic species. These transition metal carbene complexes fall into two categories:[citation needed]
- Fischer carbenes in which carbenes are tethered to a metal and an electron-withdrawing group (usually a carbonyl),
- Schrock carbenes; in which carbenes are tethered to a metal and an electron-donating group. The reactions that such carbenes participate in are very different from those in which organic carbenes participate.
Triplet state carbene chemistry edit
Persistent triplet state carbenes are likely to have very similar reactivity as other non-persistent triplet state carbenes.
Physical properties edit
Those carbenes that have been isolated to date tend to be colorless solids with low melting points. These carbenes tend to sublime at low temperatures under high vacuum.
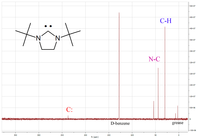
One of the more useful physical properties is the diagnostic chemical shift of the carbenic carbon atom in the 13C-NMR spectrum. Typically this peak is in the range between 200 and 300 ppm, where few other peaks appear in the 13C-NMR spectrum. An example is shown on the left for a cyclic diaminocarbene which has a carbenic peak at 238 ppm.
Upon coordination to metal centers, the 13C carbene resonance usually shifts highfield, depending on the Lewis acidity of the complex fragment. Based on this observation, Huynh et al. developed a new methodology to determine ligand donor strengths by 13C NMR analysis of trans-palladium(II)-carbene complexes. The use of a 13C-labeled N-heterocyclic carbene ligand also allows for the study of mixed carbene-phosphine complexes, which undergo trans-cis-isomerization due to the trans effect.[65]
Applications edit

NHCs are widely used as ancillary ligand in organometallic chemistry. One practical application is the ruthenium-based Grubbs' catalyst and NHC-Palladium Complexes for cross-coupling reactions.[66][67][68] NHC-metal complexes, specifically Ag(I)-NHC complexes have been widely tested for their biological applications.[69]
Preparation methods edit
NHCs are often strongly basic (the pKa value of the conjugate acid of an imidazol-2-ylidene was measured at ca. 24)[49] and react with oxygen. Clearly these reactions are performed using air-free techniques, avoiding compounds of even moderate acidity. Although imidazolium salts are stable to nucleophilic addition, other non-aromatic salts are not (i.e. formamidinium salts).[70]
In these cases, strong unhindered nucleophiles are avoided whether they are generated in situ or are present as an impurity in other reagents (such as LiOH in BuLi).
Several approaches have been developed in order to prepare stable carbenes, these are outlined below.
Deprotonation edit
Deprotonation of carbene precursor salts with strong bases has proved a reliable route to almost all stable carbenes:
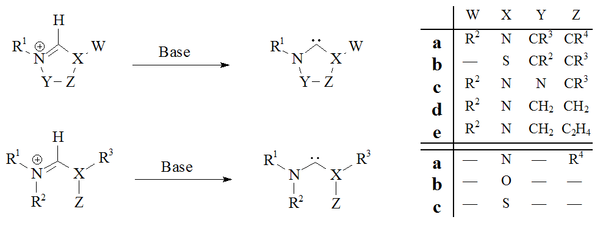
Imidazol-2-ylidenes and dihydroimidazol-2-ylidenes, such IMes, have been prepared by the deprotonation of the respective imidazolium and dihydroimidazolium salts. The acyclic carbenes[28][31] and the tetrahydropyrimidinyl[40] based carbenes were prepared by deprotonation using strong homogeneous bases.
Several bases and reaction conditions have been employed with varying success. The degree of success has been principally dependent on the nature of the precursor being deprotonated. The major drawback with this method of preparation is the problem of isolation of the free carbene from the metals ions used in their preparation.
Metal hydride bases edit
One might believe that sodium or potassium hydride[27][33] would be the ideal base for deprotonating these precursor salts. The hydride should react irreversibly with the loss of hydrogen to give the desired carbene, with the inorganic by-products and excess hydride being removed by filtration. In practice this reaction is often too slow, requiring the addition of DMSO or t-BuOH.[18][26] These reagents generate soluble catalysts, which increase the rate of reaction of this heterogeneous system, via the generation of tert-butoxide or dimsyl anion. However, these catalysts have proved ineffective for the preparation of non-imidazolium adducts as they tend to act as nucleophiles towards the precursor salts and in so doing are destroyed. The presence of hydroxide ions as an impurity in the metal hydride could also destroy non-aromatic salts.
Deprotonation with sodium or potassium hydride in a mixture of liquid ammonia/THF at −40 °C has been reported[36] for imidazole-based carbenes. Arduengo and coworkers[33] managed to prepare a dihydroimidazol-2-ylidene using NaH. However, this method has not been applied to the preparation of diaminocarbenes. In some cases, potassium tert-butoxide can be employed without the addition of a metal hydride.[26]
Alkyllithiums edit
The use of alkyllithiums as strong bases[18] has not been extensively studied, and have been unreliable for deprotonation of precursor salts. With non-aromatic salts, n-BuLi and PhLi can act as nucleophiles whilst t-BuLi can on occasion act as a source of hydride, reducing the salt with the generation of isobutene:

Amides bases edit
Lithium amides like the diisopropylamide (LDA) and the (tetramethylpiperidide (LiTMP))[28][31] generally work well for the deprotonation of all types of salts, providing that not too much LiOH is present in the n-butyllithium used to make the lithium amide. Titration of lithium amide can be used to determine the amount of hydroxide in solution. The deprotonation of precursor salts with metal hexamethyldisilazides[40] works very cleanly for the deprotonation of all types of salts, except for unhindered formamidinium salts, where this base can act as a nucleophile to give a triaminomethane adduct.
Metal-free carbene preparation edit

The preparation of stable carbenes free from metal cations has been keenly sought to allow further study of the carbene species in isolation from these metals. Separating a carbene from a carbene-metal complex can be problematic due to the stability of the complex. Accordingly, it is preferable to make the carbene free from these metals in the first place. Indeed, some metal ions, rather than stabilising the carbene, have been implicated in the catalytic dimerisation of unhindered examples.
Shown right is an X-ray structure showing a complex between a diaminocarbene and potassium HMDS. This complex was formed when excess KHMDS was used as a strong base to deprotonate the formamidinium salt. Removing lithium ions resulting from deprotonation with reagents such as lithium diisopropylamide (LDA) can be especially problematic. Potassium and sodium salt by-products tend to precipitate from solution and can be removed. Lithium ions may be chemically removed by binding to species such as cryptands or crown ethers.
Metal free carbenes have been prepared in several ways as outlined below:
Dechalcogenation edit
Another approach of preparing carbenes has relied on the desulfurisation of thioureas with potassium in THF.[29][71] A contributing factor to the success of this reaction is that the byproduct, potassium sulfide, is insoluble in the solvent. The elevated temperatures suggest that this method is not suitable for the preparation of unstable dimerising carbenes. A single example of the deoxygenation of a urea with a fluorene derived carbene to give the tetramethyldiaminocarbene and fluorenone has also been reported:[72]

The desulfurisation of thioureas with molten potassium to give imidazol-2-ylidenes or diaminocarbenes has not been widely used. The method was used to prepare dihydroimidazole carbenes.[29]
Vacuum pyrolysis edit
Vacuum pyrolysis, with the removal of neutral volatile byproducts i.e. methanol or chloroform, has been used to prepare dihydroimidazole and triazole based carbenes. Historically the removal of chloroform by vacuum pyrolysis of adducts A was used by Wanzlick[8] in his early attempts to prepare dihydroimidazol-2-ylidenes but this method is not widely used. The Enders laboratory[39] has used vacuum pyrolysis of adduct B to generate a triazol-5-ylidene.

Bis(trimethylsilyl)mercury edit
Bis(trimethylsilyl)mercury (CH3)3Si-Hg-Si(CH3)3 reacts with chloro-iminium and chloro-amidinium salts to give a metal-free carbene and elemental mercury.[73] For example:
- (CH3)3Si−Hg−Si(CH3)3 + R2N=C(Cl)−NR+
2Cl− → R2N−C−NR2 + Hg(l) + 2(CH3)3SiCl
Photochemical decomposition edit
Persistent triplet state carbenes have been prepared by photochemical decomposition of a diazomethane product via the expulsion of nitrogen gas, at a wavelength of 300 nm in benzene.
Purification edit
Stable carbenes are very reactive, and so the minimum amount of handling is desirable using air-free techniques. However, provided rigorously dry, relatively non-acidic and air-free materials are used, stable carbenes are reasonably robust to handling per se. By way of example, a stable carbene prepared from potassium hydride can be filtered through a dry celite pad to remove excess KH (and resulting salts) from the reaction. On a relatively small scale, a suspension containing a stable carbene in solution can be allowed to settle and the supernatant solution pushed through a dried membrane syringe filter. Stable carbenes are readily soluble in non-polar solvents such as hexane, and so typically recrystallisation of stable carbenes can be difficult, due to the unavailability of suitable non-acidic polar solvents. Air-free sublimation as shown right can be an effective method of purification, although temperatures below 60 °C under high vacuum are preferable as these carbenes are relatively volatile and also could begin to decompose at these higher temperatures. Indeed, sublimation in some cases can give single crystals suitable for X-ray analysis. However, strong complexation to metal ions like lithium will in most cases prevent sublimation.
References edit
- ^ Hopkinson, M. N.; Richter, C.; Schedler, M.; Glorius, F. (2014). "An Overview of N-Heterocyclic Carbenes". Nature. 510 (7506): 485–496. Bibcode:2014Natur.510..485H. doi:10.1038/nature13384. PMID 24965649. S2CID 672379.
- ^ Ronald Breslow (1957). "Mechanism of Thiamine Action: Participation of a Thiazolium Zwitterion". Chem. Ind. 26: 893.
- ^ Ronald Breslow (1958). "On the Mechanism of Thiamine Action. IV.1 Evidence from Studies on Model Systems". J. Am. Chem. Soc. 80 (14): 3719–3726. doi:10.1021/ja01547a064.
- ^ R. Breslow (1957). "Rapid Deuterium Exchange in Thiazolium Salts". J. Am. Chem. Soc. 79 (7): 1762–1763. doi:10.1021/ja01564a064.
- ^ Berkessel A.; Elfert S.; Yatham V. R.; Neudörfl J.-M.; Schlörer N. E.; Teles J. H. (2012). "Umpolung by N-Heterocyclic Carbenes: Generation and Reactivity of the Elusive 2,2-Diamino Enols (Breslow Intermediates)". Angew. Chem. Int. Ed. 51 (49): 12370–12374. doi:10.1002/anie.201205878. PMID 23081675.
- ^ Chemists Approach Elusive Breslow Intermediate Carmen Drahl
- ^ Hans-Werner Wanzlick; E. Schikora (1960). "Ein neuer Zugang zur Carben-Chemie" [A new way into carbene chemistry]. Angew. Chem. 72 (14): 494. Bibcode:1960AngCh..72..494W. doi:10.1002/ange.19600721409.
- ^ a b H. W. Wanzlick; E. Schikora (1960). "Ein nucleophiles Carben" [A nucleophilic carbene]. Chem. Ber. 94 (9): 2389–2393. doi:10.1002/cber.19610940905.
- ^ a b H. W. Wanzlick (1962). "Aspects of Nucleophilic Carbene Chemistry". Angew. Chem. Int. Ed. 1 (2): 75–80. doi:10.1002/anie.196200751.
- ^ D. M. Lemal; R. A. Lovald; K. I. Kawano (1964). "Tetraaminoethylenes. The Question of Dissociation". J. Am. Chem. Soc. 86 (12): 2518–2519. doi:10.1021/ja01066a044.
- ^ H. E. Winberg; J. E. Carnahan; D. D. Coffman; M. Brown (1965). "Tetraaminoethylenes". J. Am. Chem. Soc. 87 (9): 2055–2056. doi:10.1021/ja01087a040.
- ^ Denk M. K.; Hatano K.; Ma M. (1999). "Nucleophilic Carbenes and the Wanzlick Equilibrium A Reinvestigation". Tetrahedron Lett. 40 (11): 2057–2060. doi:10.1016/S0040-4039(99)00164-1.
- ^ Böhm Volker P. W.; Herrmann Wolfgang A. (2000). "The Wanzlick Equilibrium". Angew. Chem. Int. Ed. 39 (22): 4036–4038. doi:10.1002/1521-3773(20001117)39:22<4036::AID-ANIE4036>3.0.CO;2-L. PMID 11093196.
- ^ a b H. W. Wanzlick; H. J. Schonherr (1970). "Chemie nucleophiler Carbene, XVIII, 1) 1.3.4.5-Tetraphenyl-imidazoliumperchlorat" [Chemistry of nucleophilic carbenes, XVIII. 1) 1,3,4,5-Tetraphenylimidazolium perchlorate]. Liebigs Ann. Chem. 731: 176–179. doi:10.1002/jlac.19707310121.
- ^ R. Gleiter; R. Hoffmann (1968). "Stabilizing a singlet methylene". J. Am. Chem. Soc. 90 (20): 5457–5460. doi:10.1021/ja01022a023.
- ^ a b A. Igau; H. Grutzmacher; A. Baceiredo; G. Bertrand (1988). "Analogous α,α′-bis-carbenoid, triply bonded species: synthesis of a stable λ3-phosphino carbene-λ3-phosphaacetylene". J. Am. Chem. Soc. 110 (19): 6463–6466. doi:10.1021/ja00227a028.
- ^ a b c G. Bertrand; R. Reed (1994). "λ3-Phosphinocarbenes λ5-phosphaacetylenes". Coord. Chem. Rev. 137: 323–355. doi:10.1016/0010-8545(94)03005-B.
- ^ a b c Arduengo, A.J.; Harlow, R.L.; Kline, M. (1991). "A stable crystalline carbene". J. Am. Chem. Soc. 113 (1): 361–363. doi:10.1021/ja00001a054.
- ^ Tapu, Daniela; Dixon, David A.; Roe, Christopher (12 August 2009). "13C NMR Spectroscopy of "Arduengo-type" Carbenes and Their Derivatives". Chem. Rev. 109 (8): 3385–3407. doi:10.1021/cr800521g. PMID 19281270.
- ^ Arduengo, Anthony J.; Harlow, Richard L.; Kline, Michael (January 1991). "A stable crystalline carbene". J. Am. Chem. Soc. 113 (1): 361–363. doi:10.1021/ja00001a054.
- ^ a b A. J. Arduengo; F. Davidson; H. V. R. Dias; J. R. Goerlich; D. Khasnis; W. J. Marshall; T. K. Prakasha (1997). "An Air Stable Carbene and Mixed Carbene "Dimers"". J. Am. Chem. Soc. 119 (52): 12742–12749. doi:10.1021/ja973241o.
- ^ Christophe Buron; Heinz Gornitzka; Vadim Romanenko; Guy Bertrand (2000). "Stable Versions of Transient Push-Pull Carbenes: Extending Lifetimes from Nanoseconds to Weeks". Science. 288 (5467): 834–836. Bibcode:2000Sci...288..834B. doi:10.1126/science.288.5467.834. PMID 10796999.
- ^ Solé, Stéphane; Gornitzka, Heinz; Schoeller, Wolfgang W.; Bourissou, Didier; Bertrand, Guy (2001). "(Amino)(Aryl)Carbenes: Stable Singlet Carbenes Featuring a Spectator Substituent". Science. 292 (5523): 1901–1903. Bibcode:2001Sci...292.1901S. doi:10.1126/science.292.5523.1901. PMID 11397943.
- ^ Lai Chun-Liang; Guo Wen-Hsin; Lee Ming-Tsung; Hu Ching-Han (2005). "Ligand properties of N-heterocyclic and Bertrand carbenes: A density functional study". J. Organomet. Chem. 690 (24–25): 5867–5875. doi:10.1016/j.jorganchem.2005.07.058.
- ^ Rzepa, Henry (11 Sep 2016). "What's in a name? Carbenes: a reality check". Chemistry with a Twist. Retrieved 15 Feb 2024.
- ^ a b c d A. J. Arduengo; H. V. R. Dias; R. L. Harlow; M. Kline (1992). "Electronic stabilization of nucleophilic carbenes". J. Am. Chem. Soc. 114 (14): 5530–5534. doi:10.1021/ja00040a007.
- ^ a b c J. Arduengo; J. R. Goerlich; W. J. Marshall (1995). "A stable diaminocarbene". J. Am. Chem. Soc. 117 (44): 11027–11028. doi:10.1021/ja00149a034.
- ^ a b c d e R. W. Alder; P. R. Allen; M. Murray; A. G. Orpen (1996). "Bis(diisopropylamino)carbene". Angew. Chem. Int. Ed. 35 (10): 1121–1123. doi:10.1002/anie.199611211.
- ^ a b c d e M. K. Denk; A. Thadani; K. Hatano; A. J. Lough (1997). "Steric Stabilization of Nucleophilic Carbenes". Angew. Chem. Int. Ed. 36 (23): 2607–2609. doi:10.1002/anie.199726071.
- ^ a b c d Alder, RW; Chaker, L; Paolini, FP (2004). "Bis(diethylamino)carbene and the mechanism of dimerisation for simple diaminocarbenes". Chemical Communications (19): 2172–2173. doi:10.1039/b409112d. PMID 15467857.
- ^ a b c d e R. W. Alder; M. E. Blake (1997). "Bis(N-piperidyl)carbene and its slow dimerisation to tetrakis(N-piperidyl)ethene". Chem. Commun. (16): 1513–1514. doi:10.1039/a703610h.
- ^ a b R. W. Alder; M. E. Blake; J. M. Oliva (1999). "Diaminocarbenes; Calculation of Barriers to Rotation about Ccarbene–N Bonds, Barriers to Dimerization, Proton Affinities, and 13C NMR Shifts". J. Phys. Chem. A. 103 (50): 11200–11211. Bibcode:1999JPCA..10311200A. doi:10.1021/jp9934228.
- ^ a b c d e A. J. Arduengo, J. R. Goerlich and W. J. Marshall (1997). "A Stable Thiazol-2-ylidene and Its Dimer". Liebigs Ann. Chem. 1997 (2): 365–374. doi:10.1002/jlac.199719970213.
- ^ a b c R. W. Alder; C. P. Butts; A. G. Orpen (1998). "Stable Aminooxy- and Aminothiocarbenes". J. Am. Chem. Soc. 120 (44): 11526–11527. doi:10.1021/ja9819312.
- ^ a b Lavallo, Vincent; Canac, Yves; Donnadieu, Bruno; Schoeller, Wolfgang W.; Bertrand, Guy (2006). "Cyclopropenylidenes: From Interstellar Space to an Isolated Derivative in the Laboratory". Science. 312 (5774): 722–724. Bibcode:2006Sci...312..722L. doi:10.1126/science.1126675. PMC 2427275. PMID 16614171.
- ^ a b c W. A. Herrmann; C. Kocher; L. J. Goossen; G. R. J. Artus (1996). "Heterocyclic Carbenes: A High-Yielding Synthesis of Novel, Functionalized N-Heterocyclic Carbenes in Liquid Ammonia". Chem. Eur. J. 2 (12): 1627–1636. doi:10.1002/chem.19960021222.
- ^ a b W. A. Herrmann; M. Elison; J. Fischer; C. Kocher; G. R. J. Artus (1996). "N-Heterocyclic Carbenes: Generation under Mild Conditions and Formation of Group 8–10 Transition Metal Complexes Relevant to Catalysis". Chem. Eur. J. 2 (7): 772–780. doi:10.1002/chem.19960020708.
- ^ a b H. V. R. Dias; W. C. Jin (1994). "A stable tridentate carbene ligand". Tetrahedron Lett. 35 (9): 1365–1366. doi:10.1016/S0040-4039(00)76219-8.
- ^ a b c d D. Enders; K. Breuer; G. Raabe; J. Runsink; J. H. Teles; J. P. Melder; K. Ebel; S. Brode (1995). "Preparation, Structure, and Reactivity of 1,3,4-Triphenyl-4,5-dihydro-1H-1,2,4-triazol-5-ylidene, a New Stable Carbene". Angew. Chem. Int. Ed. 34 (9): 1021–1023. doi:10.1002/anie.199510211.
- ^ a b c d R. W. Alder; M. E. Blake; C. Bortolotti; S. Buffali; C. P. Butts; E. Lineham; J. M. Oliva; A. G. Orpen; M. J. Quayle (1999). "Complexation of stable carbenes with alkali metals". Chem. Commun. (3): 241–242. doi:10.1039/a808951e.
- ^ R. Breslow (1957). "Rapid Deuterium Exchange in Thiazolium Salts". J. Am. Chem. Soc. 79 (7): 1762–1763. doi:10.1021/ja01564a064.
- ^ a b H. D. Haztzler (1970). "Nucleophilic 1,3-dithiolium carbenes". J. Am. Chem. Soc. 92 (5): 1412–1413. doi:10.1021/ja00708a058.
- ^ a b H. D. Hartzler (1972). "1,3-Dithiolium carbenes from acetylenes and carbon disulfide". J. Am. Chem. Soc. 95 (13): 4379–4387. doi:10.1021/ja00794a039.
- ^ G. Bertrand; A. Igau; A. Baceiredo; G. Trinquier (1989). "[Bis(diisopropylamino)phosphino]trimethylsilylcarbene: A Stable Nucleophilic Carbene". Angew. Chem. Int. Ed. 28 (5): 621–622. doi:10.1002/anie.198906211.
- ^ a b Präsang, C; Donnadieu, B; Bertrand, G (2005). "Stable Planar Six-π-Electron Six-Membered N-Heterocyclic Carbenes with Tunable Electronic Properties". J. Am. Chem. Soc. 127 (29): 10182–10183. doi:10.1021/ja052987g. PMC 2440681. PMID 16028925.
- ^ Tomioka, H; Iwamoto, E; Itakura, H; Hirai, K (2001). "Generation and characterization of a fairly stable triplet carbene". Nature. 412 (6847): 626–628. Bibcode:2001Natur.412..626T. doi:10.1038/35088038. PMID 11493917. S2CID 4373216.
- ^ Michael Freemantle (2001-08-13). "Triplet Carbene has Long Life". Chemical & Engineering News. 79 (33): 11. doi:10.1021/cen-v079n033.p011a.
- ^ Itoh, T; Nakata, Y; Hirai, K; Tomioka, H (2006). "Triplet Diphenylcarbenes Protected by Trifluoromethyl and Bromine Groups. A Triplet Carbene Surviving a Day in Solution at Room Temperature". J. Am. Chem. Soc. 128 (3): 957–967. doi:10.1021/ja056575j. PMID 16417387.
- ^ a b R. W. Alder; P. R. Allen; S. J. Williams (1995). "Stable carbenes as strong bases". Chem. Commun. (12): 1267. doi:10.1039/c39950001267.
- ^ Massey Richard S (2012). "Proton Transfer Reactions of Triazol-3-ylidenes: Kinetic Acidities and Carbon Acid pKaValues for Twenty Triazolium Salts in Aqueous Solution" (PDF). J. Am. Chem. Soc. 134 (50): 20421–20432. doi:10.1021/ja308420c. PMID 23173841.
- ^ Higgins, Eleanor M.; Sherwood, Jennifer A.; Lindsay, Anita G.; Armstrong, James; Massey, Richard S.; Alder, Roger W.; O'Donoghue, Annmarie C. (2011). "pKas of the conjugate acids of N-heterocyclic carbenes in water". Chem. Commun. 47 (5): 1559–1561. doi:10.1039/C0CC03367G. PMID 21116519. S2CID 205757477.
- ^ T. A. Taton; P. Chen (1996). "A Stable Tetraazafulvalene". Angew. Chem. Int. Ed. 35 (9): 1011–1013. doi:10.1002/anie.199610111.
- ^ Alder, Roger W.; Blake, Michael E.; Chaker, Leila; Harvey, Jeremy N.; Paolini, François; Schütz, Jan (2004). "When and How Do Diaminocarbenes Dimerize?". Angew. Chem. Int. Ed. 43 (44): 5896–5911. doi:10.1002/anie.200400654. PMID 15457494.
- ^ Enders, D.; Breuer, K.; Runsink, J.; Teles, J.H. (1996). "Chemical Reactions of the Stable Carbene 1,3,4-Triphenyl-4,5-dihydro-1H-1,2,4-triazol-5-ylidene". Liebigs Ann. Chem. 1996 (12): 2019–2028. doi:10.1002/jlac.199619961212.
- ^ a b Enders, D.; Breuer, K.; Teles, J.H.; Ebel, K. (1997). "1,3,4-Triphenyl-4,5-dihydro-1H-1,2,4-triazol-5-ylidene – applications of a stable carbene in synthesis and catalysis". J. Prakt. Chem. 339: 397–399. doi:10.1002/prac.19973390170.
- ^ Wolf, J; Böhlmann, W; Findeisen, M; Gelbrich, T; Hofmann, HJ; Schulze, B (2007). "Synthesis of stable isothiazole carbenes". Angew. Chem. Int. Ed. 46 (17): 3118–3121. doi:10.1002/anie.200604305. PMID 17372997.
- ^ a b DeHope, A; Lavallo, V; Donnadieu, B; Schoeller, WW; Bertrand, G (2007). "Recently reported crystalline isothiazole carbenes: Myth or reality". Angew. Chem. Int. Ed. 46 (36): 6922–6925. doi:10.1002/anie.200702272. PMID 17661300.
- ^ Wolf Janine; Böhlmann Winfried; Findeisen Matthias; Gelbrich Thomas; Hofmann Hans-Jorg; Schulze Borbel (2007). "Reply to "Recently Reported Crystalline Isothiazole Carbenes: Myth or Reality"". Angew. Chem. Int. Ed. 46 (36): 6926. doi:10.1002/anie.200702746.
- ^ a b Wolfgang A. Herrmann; Christian Köcher (1997). "N-Heterocyclic Carbenes". Angew. Chem. Int. Ed. 36 (20): 2162–2187. doi:10.1002/anie.199721621. S2CID 97336589.
- ^ Gernot Boche; Christof Hilf; Klaus Harms; Michael Marsch; John C. W. Lohrenz (1995). "Crystal Structure of the Dimeric (4-tert-Butylthiazolato)(glyme)lithium: Carbene Character of a Formyl Anion Equivalent". Angew. Chem. Int. Ed. 34 (4): 487–489. doi:10.1002/anie.199504871.
- ^ a b D. Enders; H. Gielen; G. Raabe; J. Runsink; J. H. Teles (1996). "Synthesis and Stereochemistry of the First Chiral (Imidazolinylidene)- and (Triazolinylidene)palladium(II) Complexes". Chem. Ber. 129 (12): 1483–1488. doi:10.1002/cber.19961291213.
- ^ a b Wolfgang A. Herrmann; Martina Elison; Jakob Fischer; Christian Köcher; Georg R. J. Artus (1995). "Metal Complexes of N-Heterocyclic Carbenes – A New Structural Principle for Catalysts in Homogeneous Catalysis". Angew. Chem. Int. Ed. 34 (21): 2371–2374. doi:10.1002/anie.199523711.
- ^ a b Wolfgang A. Herrmann; Lukas J. Goossen; Christian Köcher; Georg R. J. Artus (1996). "Chiral Heterocylic Carbenes in Asymmetric Homogeneous Catalysis". Angew. Chem. Int. Ed. 35 (23–24): 2805–2807. doi:10.1002/anie.199628051.
- ^ M. Scholl; T. M. Trnka; J. P. Morgan; R. H. Grubbs (1999). "Increased ring closing metathesis activity of ruthenium-based olefin metathesis catalysts coordinated with imidazolin-2-ylidene ligands". Tetrahedron Lett. 40 (12): 2247–2250. doi:10.1016/S0040-4039(99)00217-8.
- ^ Han Vinh Huynh; et al. (2009). "13C NMR Spectroscopic Determination of Ligand Donor Strengths Using N-Heterocyclic Carbene Complexes of Palladium(II)". Organometallics. 28 (18): 5395–5404. doi:10.1021/om900667d.
- ^ S. P. Nolan [editor] (2006). N-Heterocyclic carbenes in synthesis, Wiley-VCH ISBN 3-527-31400-8
- ^ F. Glorius [editor] (2007) N-Heterocyclic carbenes in transition metal catalysis, Springer ISBN 3-540-36929-5
- ^ Díez-González, Silvia; Marion, Nicolas; Nolan, Steven P. (2009-08-12). "N-Heterocyclic Carbenes in Late Transition Metal Catalysis". Chem. Rev. 109 (8): 3612–3676. doi:10.1021/cr900074m. ISSN 0009-2665. PMID 19588961. S2CID 206902952.
- ^ Garrison Jered C.; Youngs Wiley J. (2005). "Ag(I) N-Heterocyclic Carbene Complexes: Synthesis, Structure, and Application". Chem. Rev. 105 (11): 3978–4008. doi:10.1021/cr050004s. PMID 16277368. S2CID 43090499.
- ^ Roger W. Alder; Michael E. Blake; Simone Bufali; Craig P. Butts; A. Guy Orpen; Jan Schütz; Stuart J. Williams (2001). "Preparation of tetraalkylformamidinium salts and related species as precursors to stable carbenes". J. Chem. Soc., Perkin Trans. 1 (14): 1586–1593. doi:10.1039/b104110j.
- ^ N. Kuhn; T. Kratz (1993). "Synthesis of Imidazol-2-ylidenes by Reduction of Imidazole-2(3H)-thiones". Synthesis. 1993 (6): 561–562. doi:10.1055/s-1993-25902.
- ^ D. Kovacs; M. S. Lee; D. Olson; J. E. Jackson (1996). "Carbene-to-Carbene Oxygen Atom Transfer". J. Am. Chem. Soc. 118 (34): 8144–8145. doi:10.1021/ja961324j.
- ^ Michael Otto; Salvador Conejero; Yves Canac; Vadim D. Romanenko; Valentyn Rudzevitch; Guy Bertrand (2004). "Mono- and Diaminocarbenes from Chloroiminium and -amidinium Salts: Synthesis of Metal-Free Bis(dimethylamino)carbene". J. Am. Chem. Soc. 126 (4): 1016–1017. doi:10.1021/ja0393325. PMID 14746458.
Further reading edit
Reviews on persistent carbenes:
- Hopkinson, M. N.; Richter, C.; Schedler, M.; Glorius, F. (2014). "An Overview of N-Heterocyclic Carbenes". Nature. 510 (7506): 485–496. Bibcode:2014Natur.510..485H. doi:10.1038/nature13384. PMID 24965649. S2CID 672379..
- Carbene Chemistry: From Fleeting Intermediates to Powerful Reagents, (Chapter 4, Hideo Tomioka (triplet state); Chapter 5 (singlet state), Roger W. Alder) - ed. Guy Bertrand
- Reactive Intermediate Chemistry By Robert A. Moss, Matthew Platz, Maitland Jones (Chapter 8, Stable Singlet Carbenes, Guy Bertrand)
- R. W. Alder, in 'Diaminocarbenes: exploring structure and reactivity', ed. G. Bertrand, New York, 2002
- M. Regitz (1996). "Stable Carbenes—Illusion or Reality?". Angew. Chem. Int. Ed. 30 (6): 674–676. doi:10.1002/anie.199106741.
For a review on the physico-chemical properties (electronics, sterics, ...) of N-heterocyclic carbenes:
- T. Dröge; F. Glorius (2010). "The Measure of All Rings - N-Heterocyclic Carbenes". Angew. Chem. Int. Ed. 49 (39): 6940–6952. doi:10.1002/anie.201001865. PMID 20715233.